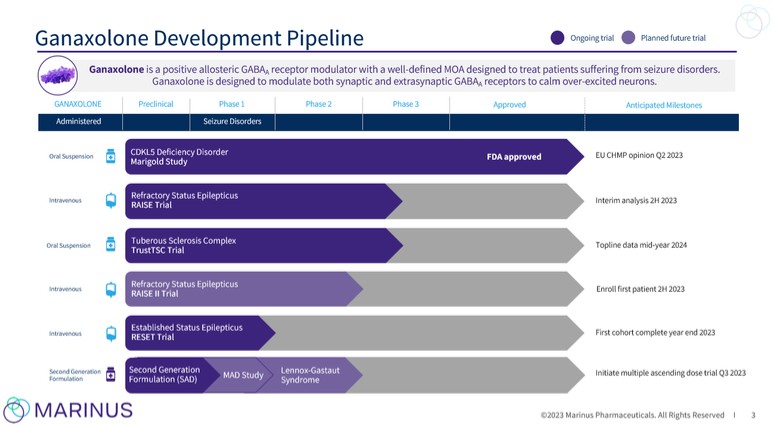
ganaxolone will depend on a variety of factors, including the uncertainties of future clinical trials and preclinical studies, uncertainties in clinical trial enrollment rate and significant and changing government regulation.
In addition, the probability of success for our clinical programs will depend on numerous factors, including competition, manufacturing capability and commercial viability. See the “Risk Factors” section of our Annual Report on Form 10-K filed on March 9, 2023 for more information with respect to such factors. Our commercial success depends upon attaining significant market acceptance, if approved, among physicians, patients, healthcare payers and the medical community. We will determine which programs to pursue and how much to fund each program in response to the scientific and clinical success, as well as an assessment of commercial potential.
Selling, General and Administrative Expenses
Selling, general and administrative expenses consist principally of salaries and related costs for executive, commercial and other administrative personnel and consultants, including stock-based compensation and travel expenses. Other selling, general and administrative expenses include professional fees for commercial, legal, patent review, consulting and accounting services. Selling, general and administrative expenses are expensed when incurred.
Cost of Product Revenue
Cost of product revenue includes the cost of inventory sold, which includes direct manufacturing and supply chain costs. Also included in cost of product revenue are royalty payments owed to Purdue Neuroscience Company (Purdue) and Ovid in accordance with the respective license agreements. We began capitalizing inventory related to ZTALMY subsequent to the March 2022 FDA approval of ZTALMY, as the related costs were expected to be recoverable through the commercialization and subsequent sale of ZTALMY. Prior to FDA approval of ZTALMY, costs estimated at approximately $2 million for commercially saleable product and materials were incurred and included in research and development expenses. As a result, cost of product revenues related to ZTALMY will initially reflect a lower average per unit cost of materials into approximately the first half of 2024, as previously expensed inventory is utilized for commercial production and sold to customers.
Cost of IP License Fee
In March 2022, we entered into an exclusive patent license agreement (License Agreement) with Ovid. Under the License Agreement, we have an exclusive, non-transferable (except as provided in the License Agreement), royalty-bearing, sublicensable license under certain of Ovid’s patent(s) and patent applications to develop, make, have made, commercialize, promote, distribute, sell, offer for sale and import, ganaxolone, including any analogues or derivatives, including its salts, and pharmaceutical formulations of the foregoing (Licensed Products), in the U.S., the member states of the EU, Iceland, Lichtenstein, Norway, the United Kingdom, and Switzerland (Territory) for the treatment of CDD in humans (Field). Under the License Agreement, we have the sole right and responsibility for, and control over, all development, manufacturing, and commercialization activities, including all regulatory activities, with respect to the Licensed Products in the Field in the Territory. In addition, all regulatory approvals and related filings with respect to the Licensed Products in the Field in the Territory will be in the name of, and be owned solely by, us. We were required, at Ovid’s option exercisable in accordance with the License Agreement, to (i) pay to Ovid the sum of $1.5 million in cash; or (ii) issue to Ovid 123,255 shares of our common stock, which option to obtain shares of our common stock was exercisable within the five-business day period following the filing of our Annual Report on Form 10-K for the year ended December 31, 2021 on March 24, 2022. On March 29, 2022, we issued 123,255 shares of our common stock to Ovid, per Ovid’s option in accordance with the License Agreement. As such, we recorded $1.2 million of IP license fee expenses related to the Ovid License Agreement in the three months ended March 31, 2022.
The License Agreement also provides for payment of royalties by us to Ovid in the low single digits on net sales by us, our affiliates and sublicensees, of Licensed Products in the Field in the Territory. Such royalties are subject to reduction in the event of generic competition in accordance with the License Agreement. We may terminate the License Agreement at any time without cause on thirty days’ prior written notice. Either party may terminate the License Agreement for the other party’s material breach or insolvency subject to certain cure periods. Also, Ovid has the right to terminate the License Agreement if there has not been a first commercial sale of any Licensed Products in the Field in