UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |
For the fiscal year ended December 31, 2019
or
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 | |
For the transition period from to
Commission File Number: 000-50679
(Exact Name of Corporation as Specified in Its Charter)
(State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) |
(Address of principal executive offices) (zip code)
(650 ) 327-3270
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12 (b) of the Act:
Title of each class | Trading Symbol(s) | Name of each exchange on which registered |
Securities registered pursuant to Section 12 (g) of the Act:
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Acts. Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports); and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.:
☒ | Accelerated filer | ☐ | ||
Non-accelerated filer | ☐ | Smaller reporting company | ||
Emerging growth company | ||||
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ☐ No ☒
The aggregate market value of voting and non-voting common equity held by non-affiliates of the Registrant as of June 30, 2019 was $1,071,264,355 , based on the closing price of $11.15 for shares of the Registrant’s common stock as reported on the Nasdaq Stock Market on June 28, 2019, the last trading day before June 30, 2019. Shares of common stock beneficially owned by each executive officer, director and holder of more than 10% of our common stock have been excluded, in that such persons may be deemed to be affiliates. This calculation does not reflect a determination that certain persons are affiliates of the Registrant for any other purpose.
On February 12, 2020 there were 114,594,745 shares of common stock outstanding at a par value of $0.001 per share.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the Registrant’s definitive proxy statement for its 2020 Annual Meeting of Stockholders are incorporated by reference in Items 10, 11, 12, 13 and 14 of Part III.
TABLE OF CONTENTS
Form 10-K
For the year ended December 31, 2019
Page | ||||
PART I | ||||
ITEM 1. | ||||
ITEM 1A. | ||||
ITEM 1B. | ||||
ITEM 2. | ||||
ITEM 3. | ||||
ITEM 4. | ||||
PART II | ||||
ITEM 5. | ||||
ITEM 6. | ||||
ITEM 7. | ||||
ITEM 7A. | ||||
ITEM 8. | ||||
ITEM 9. | ||||
ITEM 9A. | ||||
ITEM 9B. | ||||
PART III | ||||
ITEM 10. | ||||
ITEM 11. | ||||
ITEM 12. | ||||
ITEM 13. | ||||
ITEM 14. | ||||
PART IV | ||||
ITEM 15. | ||||
ITEM 16. | ||||
PART I
This Annual Report on Form 10-K (“Form 10-K”) contains forward-looking statements within the meaning of Section 21E of the Securities Exchange Act of 1934, as amended (“Exchange Act”), and Section 27A of the Securities Act of 1933, as amended (“Securities Act”). All statements contained in this Form 10-K, other than statements of historical fact, are forward-looking statements. When used in this report, the words “believe,” “anticipate,” “intend,” “plan,” “estimate,” “expect,” “may,” “will,” “should, “would,” “could,” “seek” and similar expressions are forward-looking statements based on management’s current expectations. The absence of these words does not mean that a statement is not forward-looking. Forward-looking statements include, but are not limited to, statements about:
• | our ability to manufacture, market and sell Korlym® (mifepristone) 300 mg Tablets (“Korlym”); |
• | our estimates regarding enrollment in and the completion dates of our clinical trials and the anticipated results of these trials; |
• | the progress and timing of our research and development programs and the regulatory activities associated with them; |
• | our ability to realize the benefits of orphan drug designation for Korlym and the impact of possible future competition for Korlym or our product candidates; |
• | our estimates for future performance, including revenue and profits; |
• | the timing of the market introduction of future product candidates, including new uses for Korlym and any of our proprietary selective cortisol modulators; |
• | our ability to manufacture, market, commercialize and achieve market acceptance for our product candidates; |
• | uncertainties associated with obtaining and enforcing patents; and |
• | estimates regarding our capital requirements. |
Forward-looking statements involve risks and uncertainties and are not guarantees of future performance. Actual events or results may differ materially for many reasons. For a more detailed discussion of the risks and uncertainties that may affect the accuracy of our forward-looking statements, see the “Risk Factors,” “Overview” and “Liquidity and Capital Resources” sections of the “Management’s Discussion and Analysis of Financial Condition and Results of Operations” sections of this Form 10-K. You should also carefully consider the other reports and documents we file with the Securities and Exchange Commission (“SEC”).
Forward-looking statements in this Form 10-K reflect our view only as of the date of this report. Except as required by law, we undertake no obligation to update forward-looking statements.
Unless stated otherwise, all references in this document to “we,” “us,” “our,” “Corcept,” the “Company,” “our company” and similar words and phrases refer to Corcept Therapeutics Incorporated.
ITEM 1. BUSINESS
Overview
We are a commercial-stage company engaged in the discovery and development of drugs that treat severe metabolic, oncologic and psychiatric disorders by modulating the effects of the hormone cortisol. Since 2012, we have marketed Korlym® (mifepristone) for the treatment of patients who suffer from Cushing’s syndrome, a disease caused by excess cortisol activity.
We have discovered more than 500 proprietary, selective cortisol modulators in four structurally distinct series. These novel molecules share Korlym’s affinity for the glucocorticoid receptor (“GR”) but, unlike Korlym, do not bind to the progesterone receptor (“PR”) and therefore do not cause effects arising from antagonism of progesterone activity, such as termination of pregnancy, endometrial thickening and vaginal bleeding. The composition of these compounds and their methods of use in a wide range of indications are covered by U.S. and foreign patents. Our lead compounds have entered the clinic as potential treatments for a variety of serious disorders - Cushing’s syndrome, solid tumors (including advanced, high-grade serous ovarian cancer, metastatic pancreatic cancer and castration-resistant prostate cancer), weight gain caused by antipsychotic medications, and non-alcoholic steatohepatitis (“NASH”).
1
The Role of Cortisol in Disease
Cortisol is a steroid hormone that plays a significant role in how the body reacts to stress. It is essential for survival. Cortisol influences metabolism and the immune system and contributes to emotional stability. Cortisol levels follow a diurnal rhythm that is essential to health, peaking upon awakening and decreasing during the day. Insufficient cortisol activity may lead to dehydration, hypotension, shock, fatigue and hypoglycemia. Excessive cortisol activity, known as hypercortisolism, may lead to a suppressed immune response, impaired glucose tolerance, diabetes, obesity, fatty liver disease, depressed mood, psychosis, wasting of the arms and legs, edema, fatigue, hypertension and other problems. Pre-clinical and clinical data suggest that cortisol reduces a patient’s immune response to oncogenesis, shields certain cancer cells from the apoptotic effects of chemotherapy and facilitates the growth of others.
The challenge in treating a patient with hypercortisolism is modulating cortisol’s effects without suppressing them below normal levels or disrupting cortisol’s normal diurnal rhythm. Simply reducing or destroying the ability of the body to make cortisol can cause serious harm. Cortisol activity can be modulated effectively by a drug that competes with cortisol as it attempts to bind to GR. Mifepristone, the active ingredient in Korlym, is a competitive GR antagonist, as are our proprietary compounds.
Because mifepristone works by reducing the binding of excess cortisol to GR, it can modulate the effects of abnormal levels and release patterns of cortisol without compromising cortisol’s healthy functions and rhythms. However, mifepristone also binds to PR, thereby terminating pregnancy and causing other adverse effects, including vaginal bleeding (a debilitating condition suffered by a significant portion of women who take Korlym). Our proprietary selective cortisol modulators bind to GR as potently as mifepristone does, but have no affinity for PR and so do not cause PR-related side effects.
Cushing’s Syndrome
Background. Cushing’s syndrome is the clinical manifestation of hypercortisolism. An estimated 10 to 15 of every one million people are diagnosed with Cushing’s syndrome each year, resulting in approximately 3,000 new patients and a patient population in the United States of about 20,000, approximately half of whom are cured by surgery. Cushing’s syndrome most often affects adults between the ages of 20 and 50.
Most people with Cushing’s syndrome have one or more of the following symptoms: high blood sugar, diabetes, high blood pressure, upper body obesity, rounded face, increased fat around the neck, thinning arms and legs, severe fatigue and weak muscles. Irritability, anxiety, cognitive disturbances and depression are also common. Cushing’s syndrome can affect every organ system in the body and can be lethal if not treated. The preferred treatment is surgery, which, if successful, can cure the disease. In approximately half of the patients, surgery is not successful because the tumor cannot be located or removed completely. Depending on the type of tumor, surgery can also result in a range of complications.
Korlym to Treat Patients with Cushing’s Syndrome. We sell Korlym exclusively in the United States, using experienced sales representatives to call on physicians caring for patients with endogenous Cushing’s syndrome (hypercortisolism). Because many people who suffer from Cushing’s syndrome are undiagnosed or inadequately treated, we have developed and continue to refine and expand programs to educate physicians and patients about screening for hypercortisolism and the role Korlym can play in treating the disorder. We also have a field-based force of medical science liaisons.
We use one specialty pharmacy and one specialty distributor to distribute Korlym and provide logistical support to physicians and patients. Our policy is that no patient with Cushing’s syndrome will be denied access to Korlym for financial reasons. To help us achieve that goal, we fund our own patient support programs and donate money to independent charitable foundations that help patients pay for all aspects of their Cushing’s syndrome care, whether or not that care includes taking Korlym.
Relacorilant to Treat Patients with Cushing’s Syndrome. We are conducting a Phase 3 trial of our proprietary, selective cortisol modulator, relacorilant, as a potential treatment for hypercortisolism. Relacorilant was well-tolerated in its Phase 1 and Phase 2 trials. Patients in the Phase 2 trial exhibited meaningful improvements in glucose control and hypertension, as well as weight loss, improved liver function, coagulopathy, cognition, mood, insulin resistance, and quality of life. Importantly, relacorilant shares Korlym’s affinity for GR, but, unlike Korlym, has no affinity for PR, and so does not cause the effects associated with PR affinity, including termination of pregnancy, endometrial thickening and vaginal bleeding. Relacorilant also does not appear to cause hypokalemia (low potassium), a potentially serious adverse event that is the leading cause of patients stopping treatment with Korlym. Forty-four percent of patients in Korlym’s pivotal trial experienced hypokalemia.
Relacorilant’s Phase 3 trial (“GRACE”), is expected to enroll 130 patients at sites in the United States, Canada, Europe and Israel. Each patient in GRACE will receive relacorilant for 22 weeks. Those who exhibit pre-specified improvements in hypertension or glucose metabolism will then enter a 12-week, double-blind, “randomized withdrawal” phase, in which half of the patients will continue receiving relacorilant and the rest will receive placebo. GRACE’s primary endpoints are the rate and degree of relapse in patients receiving placebo compared to those continuing treatment with relacorilant.
2
We also plan to conduct a placebo-controlled, double-blind, Phase 3 trial of relacorilant to treat patients whose Cushing’s syndrome is caused by an adrenal tumor. This etiology of Cushing’s syndrome has not been rigorously studied. Patients with adrenal Cushing’s syndrome have poor health outcomes and would benefit from an improved understanding of the role cortisol modulation may play in their treatment.
The United States Food and Drug Association ("FDA") and the European Commission (“EC”) have designated relacorilant as an orphan drug for the treatment of Cushing’s syndrome. In the United States, orphan designation confers tax credits, reduced regulatory fees and, provided we obtain approval for the treatment of Cushing's syndrome, 7 years of exclusive marketing rights for relacorilant in the treatment of Cushing’s syndrome, with limited exceptions. Benefits of orphan drug designation by the EC are similar, and include reduced regulatory fees and, if we obtain approval, ten years of exclusive marketing rights in the European Union (“EU”) for the treatment of Cushing’s syndrome. Additional benefits in the EU include protocol assistance from the European Medicines Agency (“EMA”) and access to the EU's centralized marketing authorization procedure. The EC based its orphan designation on its finding that there was plausible evidence of relacorilant’s efficacy and potential to confer significant clinical benefit compared to already-approved treatments.
In neither the United States nor the EU does orphan drug designation shorten the drug approval process, make approval more likely or prevent competitors from marketing other drugs for the treatment of Cushing’s syndrome.
FKBP5 Gene Expression Assay. The tests used to diagnose patients with hypercortisolism and optimize their treatment are imprecise and often fail to identify patients with less severe manifestations of the disease. We have developed an assay to measure expression of the gene FKBP5, which is stimulated by cortisol activity, and have completed analytical validation pursuant to the Clinical Laboratory Improvement Amendments (“CLIA”). Clinical data indicate that FKBP5 levels are high in patients suffering from hypercortisolism (i.e., excess cortisol activity), but subside when they are successfully treated. We are testing this hypothesis in the GRACE trial. We believe successful development of this assay will enable physicians to identify new patients with hypercortisolism more easily and to better treat those already in their care.
Oncology
There is substantial in vitro, in vivo and clinical evidence that cortisol’s activity allows certain solid tumors to resist treatment. In some cancers, cortisol activity promotes tumor growth. In other cancers, cortisol stimulates genes that retard cellular apoptosis. Cortisol also suppresses the body's immune response. However, activating, not suppressing, the immune system is beneficial in fighting certain cancers. Adding a cortisol modulator to a treatment regimen may help the patient’s immune system combat the disease. Many types of solid tumors express GR and are potential targets for cortisol modulation therapy, among them pancreatic, ovarian, castration-resistant prostate and adrenocortical cancer. We own, or have exclusively licensed, several patents covering the use of cortisol modulators to treat pancreatic, cervical, breast, and prostate cancers.
Relacorilant in Patients with Solid Tumors. At the June 2019 annual meeting of the American Society of Clinical Oncology (“ASCO”), we presented data from our Phase 1/2 trial of relacorilant plus nab-paclitaxel (Celgene Corporation’s Abraxane®) to treat patients with advanced solid tumors. Eleven of the response-evaluable patients in that trial suffered from advanced, high-grade serous ovarian cancer. Five of these patients experienced disease control of 16 weeks or greater. Of the trial’s 25 response-evaluable patients with pancreatic tumors, seven had disease control of 16 weeks or greater.
These are positive results in such ill patients, particularly in patients who had received prior taxane-based treatment, and merit further investigation. A Phase 2, controlled trial of relacorilant in combination with Abraxane in patients with advanced, high-grade serous ovarian tumors is ongoing. The trial is expected to enroll 180 patients at sites in the United States and Europe. Two-thirds of the patients will receive relacorilant plus Abraxane. The rest will receive Abraxane alone. The primary endpoint is progression-free survival (“PFS”), as measured using the Response Evaluation Criteria in Solid Tumors ("RECIST").
We plan to conduct a Phase 3 trial of relacorilant plus Abraxane to treat patients with metastatic pancreatic cancer. Relacorilant has been designated an orphan drug by both the FDA and the EC for the treatment of pancreatic cancer.
We own or have exclusively licensed U.S. and European patents covering relacorilant’s composition of matter and its use to treat a variety of disorders, including pancreatic cancer, castration-resistance prostate cancer ("CRPC") and other solid tumors.
Cortisol Modulators to Treat Patients with CRPC. Because androgens stimulate prostate tumor growth, androgen deprivation is a common treatment for metastatic prostate cancer. Tumors eventually escape androgen deprivation therapy through the proliferation of cells for which cortisol’s stimulation of GR and cortisol’s stimulation of mutated androgen receptors are primary growth factors. Combining a cortisol modulator with an androgen modulator such as Xtandi may block this escape route.
We are conducting a dose-finding trial of our proprietary, selective cortisol modulator exicorilant combined with Xtandi in patients with metastatic CRPC. Investigators at the University of Chicago are conducting a dose-finding trial of relacorilant
3
combined with Xtandi in the same patient population. We are providing relacorilant. In addition to patents covering its composition of matter, we own U.S. patents covering the use of exicorilant to treat CRPC.
Antipsychotic-Induced Weight Gain and NASH
In animal models, our proprietary selective cortisol modulator miricorilant potently prevents and reverses the weight gain caused by Eli Lilly and Company’s antipsychotic medication Zyprexa® (olanzapine). These findings are similar to the results generated with mifepristone in the same animal models and from placebo-controlled clinical trials in which mifepristone significantly reduced the weight gain and adverse metabolic effects experienced by healthy subjects administered Zyprexa or Johnson & Johnson’s antipsychotic medication Risperdal® (risperidone). The results of the clinical trials were published in the journals Advances in Therapy, Gross et al (2009) and Obesity, Gross et al (2010).
We are conducting a double-blind, placebo-controlled Phase 1b trial testing miricorilant's activity in attenuating antipsychotic-induced weight gain. We have completed the first part of this trial, which enrolled 66 healthy subjects, each of whom received ten mg per day of olanzapine and either placebo or miricorilant (600 mg) for 14 days.
The average weight gain on day eight was 3.5 kilograms in subjects who received olanzapine plus placebo, compared to 2.6 kilograms in those who received olanzapine plus miricorilant (p=0.04). On Day 15, the placebo group gained an average of 5.0 kilograms while the miricorilant group gained 3.9 kilograms (p=0.01). Markers of liver damage that often rise temporarily upon initiation of olanzapine increased less in subjects receiving miricorilant. On Day 12, the enzyme alanine aminotransferase (ALT) increased 144.5 IU/L in the placebo group compared to 111.3 IU/L in the miricorilant group (p=0.11). A similar result was measured with respect to aspartate transaminase (AST), which increased 67.2 IU/L in the placebo group but only 43.3 IU/L in the miricorilant group (p=0.02). Miricorilant was well-tolerated.
The trial’s second stage is testing miricorilant dose of 900 mg. Planned enrollment is 30 healthy subjects.
We are also conducting a Phase 2, double-blind, placebo-controlled trial of miricorilant in the reversal of antipsychotic-induced weight gain. We expect to enroll 100 patients with schizophrenia at 20 sites in the United States. Study participants will continue to receive their established antipsychotic medication and will have either miricorilant (600 mg) or placebo added to their regimen for 12 weeks. The trial’s primary endpoint is reduction in weight.
Miricorilant is also potent in animal models of fatty liver and liver fibrosis, precursors of NASH, a serious disorder that afflicts millions of people in the United States. We plan to conduct a double-blind, placebo-controlled Phase 2 trial evaluating miricorilant as a treatment for NASH.
Development of our Other Selective Cortisol Modulators
Our portfolio of proprietary selective cortisol modulators, which includes relacorilant, exicorilant and miricorilant, consists of more than 500 compounds in four structurally distinct series. These compounds potently bind to GR but not the progesterone, estrogen or androgen receptors. Many of them have demonstrated positive results in animal or in vitro models of cortisol modulation. We plan to continue identifying and developing proprietary, selective cortisol modulators. We hold U.S. and foreign patents covering these compounds and their methods of use in a wide range of indications. We have applied, and will continue to apply, for patents covering the composition and method of use of our products and product candidates. See “Business – Intellectual Property.”
Studies by Independent Investigators
For many years we have advanced our understanding of cortisol modulation by supporting the work of independent academic investigators. These researchers have studied the utility of mifepristone or our proprietary selective cortisol modulators in a wide range of disorders, including central serous retinopathy, post-traumatic stress disorder, anxiety, alcoholism, cocaine addiction, Alzheimer’s disease, ALS, Cushing’s syndrome, metabolic syndrome, atherosclerosis, fatty liver disease, and solid tumors, including triple-negative breast, prostate, ovarian and non-small cell lung cancers, as well as sarcoma and melanoma.
Clinical Trial Agreements
Our clinical trials are conducted through the use of clinical research organizations (“CROs”). Our Phase 3 GRACE trial of relacorilant for the treatment of patients with Cushing’s syndrome is being conducted under an agreement with ICON plc (“ICON”). IQVIA (formerly, "Novella Clinical LLC") is helping us conduct our Phase 2 trial of relacorilant to treat patients with metastatic ovarian cancer and our dose-finding trial of exicorilant to treat patients with CRPC. Medpace, Inc. ("Medpace") is helping us conduct our Phase 2 trial testing miricorilant's activity in reversing recent antipsychotic-induced weight gain. Our agreements with ICON and IQVIA may be terminated by us on 60 days’ written notice or sooner if the parties mutually agree. Our agreement with Medpace may be terminated by us without cause at any time.
4
Research and Development Spending
We incurred $89.0 million, $75.2 million and $40.4 million of research and development expenses in the years ended December 31, 2019, 2018 and 2017, respectively, which accounted for 46 percent, 47 percent and 38 percent, respectively, of our total operating expenses in those years.
Manufacturing Korlym
We do not have manufacturing capabilities and rely on experienced contract manufacturers to produce Korlym and our product candidates. In March 2014, we entered into an agreement with Produits Chimiques Auxiliaires et de Synthese SA (“PCAS”) to produce mifepristone, the active pharmaceutical ingredient (“API”) in Korlym. In 2018, we amended this agreement and extended its term to December 31, 2021, with two one-year renewals that will occur automatically unless either party gives 12 months advance written notice of its intent not to renew. The amendment also provides for exclusivity between PCAS and Corcept, unless PCAS is unable to meet our requirements, in which case we may purchase mifepristone from another supplier.
We have agreements with two third-party manufacturers to produce and bottle Korlym tablets.
Hatch-Waxman Amendments to the Federal Food, Drug and Cosmetic Act (“FDCA”)
The FDCA establishes an approval process for generic versions of approved drugs (“Innovator Drugs”) through the submission of an Abbreviated New Drug Application (“ANDA”). An ANDA provides for marketing of a generic drug with the same active ingredients, dosage form, strength, route of administration, labeling, performance characteristics and intended use, among other things, to the Innovator Drug. ANDAs are termed “abbreviated” because they are generally not required to include preclinical and clinical data establishing safety and efficacy. Instead, generic applicants must demonstrate that their product is bioequivalent to, or performs in the same manner as, the Innovator Drug.
In seeking approval, ANDA applicants must certify to the FDA that any Orange Book patents relating to the Innovator Drug are invalid or will not be infringed by the manufacture, use or sale of the generic product. This is known as a “Paragraph IV certification.” If the owner of the Innovator Drug responds to receipt of a paragraph IV certification by suing the ANDA applicant for patent infringement, the FDA may not approve the ANDA application until the earlier of 30 months or when the trial of any infringement case concerning each such patent is favorably decided in the ANDA applicant's favor or settled, or such shorter or longer period as may be ordered by a court. This prohibition is generally referred to as the “30-month stay.” Owners of Innovator Drugs regularly challenge paragraph IV certifications and trigger 30-month stays, recognizing that the related patent litigation may take many months or years to resolve.
We are engaged in ANDA litigation with Teva Pharmaceuticals USA, Inc. ("Teva") and Sun Pharmaceutical Industries Limited (“Sun Ltd.”). In addition, Teva has challenged the validity of one of our patents in a post grant review ("PGR") proceeding before the Patent Trial and Appeal Board ("PTAB"). See "Part I, Item 3, Legal Proceedings."
Competition for Korlym
Korlym competes with established treatments, including surgery, radiation and other medications, including “off-label” uses of drugs such as ketoconazole, an anti-fungal medication. Korlym also competes with Signifor® (pasireotide) Injection, a drug marketed by the Italian pharmaceutical company Recordati S.p.A. ("Recordati") The FDA approved Signifor in December 2012 for the treatment of adult patients with Cushing’s disease who are not candidates for pituitary surgery or for whom surgery did not work. Cushing’s disease is a subset of Cushing’s syndrome.
The orphan drug marketing exclusivity period for Korlym ended in February 2019, which means a competitor that receives FDA approval for a generic equivalent of Korlym may market its drug to patients with Cushing’s syndrome, provided doing so would not infringe any of our patents. Korlym may also experience competition from generic versions and from new compounds. For example, Strongbridge Biopharma plc is conducting Phase 3 trials of levoketoconazole, a chiral form of the cortisol synthesis inhibitor ketoconazole in the United States and Europe. Recordati is developing the cortisol synthesis inhibitor osilodrostat. In November 2019, the Committee for Medicinal Products for Human Use recommended that osilodrostat be approved in the EU for the treatment of endogenous Cushing’s syndrome. Recordati has announced that it also plans to seek marketing approval for osilodrostat in the United States.
5
Intellectual Property
Patents and other proprietary rights are important to our business. We own ten composition of matter patents covering our selective cortisol modulators and 44 patents covering the use of cortisol modulators to treat a variety of serious disorders, including Cushing’s syndrome. We have exclusively licensed seven method of use patents from the University of Chicago and own an extensive portfolio of patents in countries around the world. We have applied, and will continue to apply, for U.S. and foreign patents covering the composition and method of use of our products and product candidates.
Korlym. The composition of matter patent covering Korlym’s active ingredient, mifepristone, has expired. The only other FDA-approved use of mifepristone is to terminate pregnancy. We hold 12 method of use patents listed in the FDA Orange Book covering various uses of Korlym in the treatment of patients with Cushing’s syndrome, with additional patent applications that may be suitable for listing in the Orange Book under examination at the USPTO. Our current Orange Book patents have expiration dates ranging from 2028 to 2037.
To protect our market for Korlym we rely on (1) our method of use patents, (2) the significant restrictions imposed by the FDA on the use of mifepristone to terminate pregnancy and (3) the different patient populations, administering physicians and treatment settings between the use of mifepristone to terminate pregnancy and to treat Cushing’s syndrome.
Oncology. We have exclusively licensed seven method of use patents from the University of Chicago covering the use of glucocorticoid receptor antagonists, including mifepristone, in the treatment of castration-resistant prostate cancer in combination with androgen deprivation agents and triple-negative breast cancer in combination with anti-cancer agents. See “Business - License Agreements.”
Other Method of Use Patents. In addition to our patents relating to Cushing’s syndrome, we own U.S. and foreign patents for the use of cortisol modulators in the treatment of pancreatic cancer, weight gain caused by antipsychotic medications, mild cognitive impairment, delirium, catatonia, psychosis induced by interferon-alpha therapy, migraine headaches, gastroesophageal reflux disease, neurological damage in premature infants and in the treatment of diseases using combination steroid and GR antagonist therapy. We own patents covering the optimization of mifepristone plasma levels in the treatment of patients suffering from disorders, including Cushing’s syndrome, amenable to treatment with mifepristone. We also own patents covering prevention and treatment of stress disorders, improvement of therapeutic response to electroconvulsive therapy and inhibition of cognitive deterioration in adults with Down’s Syndrome. The expiration dates of these patents and their foreign counterparts range from 2020 to 2038.
Composition of Matter Patents Covering Our Proprietary, Selective Cortisol Modulators. We have ten U.S. composition of matter patents containing claims relating to our next-generation cortisol modulators. Four of these patents have issued in Europe. The expiration dates of these patents and their foreign counterparts range from 2026 to 2033.
We have filed, in appropriate jurisdictions, foreign patent applications corresponding to our U.S. patents and applications. We cannot assure you that any of our patent applications will result in the issuance of patents, that any issued patent will include claims of the breadth we are seeking, or that competitors or other third-parties will not successfully challenge or circumvent our patents if they are issued.
We believe our patents are valid and do not infringe the patents or other proprietary rights of others. Accordingly, we believe we are not obligated to pay royalties relating to the use of intellectual property to any third parties except the University of Chicago, from which we have licensed certain patents.
License Agreements
We have exclusively licensed from the University of Chicago seven U.S. patents for (a) the use of cortisol modulators in the treatment of triple-negative breast cancer, and (b) the use of cortisol modulators to treat castration-resistant prostate cancer. We are required to pay the University of Chicago customary milestone fees and royalties on revenue from products commercialized under the issued patents or patents that may issue pursuant to the pending applications. Our license will end upon expiration of the licensed patents in 2031 and 2033 or upon notification by us to the University of Chicago. Three patents licensed from Stanford University expired in October 2018. See “Business – Intellectual Property.”
Government Regulation
Prescription pharmaceutical products are subject to extensive pre- and post-approval regulation governing the testing, manufacturing, safety, efficacy, labeling, storage, record keeping, advertising and promotion of the products under the Federal Food, Drug and Cosmetic Act. All of our product candidates require regulatory approval by government agencies prior to commercialization and are subject to continued regulatory oversight thereafter. Before a new drug may be marketed in the United States the FDA generally requires the following: completion of preclinical laboratory and animal testing; submission of an
6
Investigational New Drug (“IND”), which must become effective before clinical trials may begin; performance of adequate and well-controlled human clinical trials to establish the safety and efficacy of the proposed drug’s intended use; and approval by the FDA. Complying with these and other federal and state statutes and regulations involves significant time and expense.
Preclinical studies are generally conducted in laboratory animals to evaluate the potential safety and the efficacy of a product. Drug developers submit the results of preclinical studies to the FDA as a part of an IND, which the FDA must approve before beginning clinical trials in humans. If the clinical trial will be conducted in Europe, a Clinical Trial Authorization must be submitted and approved by the appropriate European regulatory agency prior to the commencement of the study. Typically, human clinical trials are conducted in three sequential phases that may overlap.
• | Phase 1. The product candidate is administered to a small number of healthy subjects to provide preliminary information as to its safety, tolerability and pharmacokinetics and sometimes to provide preliminary information as to its activity and/or efficacy. |
• | Phase 2. The product candidate is administered to patients afflicted with the target disease to determine its preliminary efficacy, optimal dosages and to provide more evidence of safety. |
• | Phase 3. The product candidate is administered to a larger group of patients afflicted with the target disease to establish its risk/benefit ratio and to demonstrate with substantial evidence its efficacy and safety. |
The FDA and the institutional review boards associated with clinical trial sites closely monitor the progress of clinical trials conducted in the United States and may reevaluate, alter, suspend or terminate a trial at any time for various reasons, including a belief that the subjects are being exposed to unacceptable risks. The FDA may also require that additional trials be conducted.
After Phase 3 trials are completed, drug developers submit the results of preclinical studies, clinical trials, formulation studies and data supporting manufacturing to the FDA in the form of a New Drug Applications (“NDA”). The FDA reviews an NDA upon submission and may request additional information rather than accept an NDA for filing. If the FDA accepts an NDA for filing, it may grant marketing approval (i.e., permit commercial sales), request additional information or deny the application. Once an NDA has been accepted for filing, by law the FDA has 180 days to examine the application and respond to the applicant. However, the review process is often significantly extended by FDA requests for additional information or clarification. Under the Prescription Drug User Fee Act, the FDA has a goal of responding to NDAs within ten months of the filing date for standard review, and six months for priority review, which the FDA may undertake, in its sole discretion, if a sponsor shows that its drug candidate is designed to treat a serious condition, and if approved, would provide a significant improvement in safety or effectiveness compared to marketed drugs. FDA approvals may not be granted on a timely basis or at all.
If the FDA approves a NDA, physicians may prescribe the subject drug to patients in the United States. The FDA may withdraw a product’s marketing approval if compliance with regulatory standards is not maintained. The drug developer must submit periodic reports to the FDA. Adverse patient experiences with the product must be reported to the FDA, which could result in the imposition of marketing restrictions through labeling changes or removal of the product from the market. In addition, the FDA may require post-marketing studies, referred to as “Phase 4 studies,” to monitor or further explore the effect of approved products, and may limit further marketing of the product based on the results of such studies.
Facilities involved in the manufacture of drugs are subject to periodic inspection by the FDA and other regulatory authorities and must comply with FDA-mandated current Good Manufacturing Practices regulations (“cGMP”). Failure to comply with statutory and regulatory requirements subjects the manufacturer to possible legal or regulatory action, including suspension of manufacturing or a product recall.
The FDA imposes complex regulations regarding the promotion and sale of pharmaceuticals, including standards for direct-to-consumer advertising, off-label promotion, and industry-sponsored scientific and educational activities. Failure to abide by these regulations can result in penalties including the issuance of a warning letter directing a company to correct deviations from FDA regulations, mandated modification of promotional materials and labeling and the issuance of corrective information in addition to state and federal civil and criminal penalties.
A drug developer may conduct preclinical and clinical trials investigating the use of an approved drug for the treatment of other, unapproved indications. FDA approval is required before the drug can be marketed for these indications.
Marketing Approvals Outside the United States
If we choose to distribute our product candidates outside the United States, we (or our potential future partners) will have to complete an approval process similar to the one imposed by the FDA. The approval procedure and the time required for approval vary from country to country and may involve additional preclinical and clinical trials. Foreign approvals may not be granted on a timely basis, or at all. Regulatory approval of pricing is required in most countries other than the United States, which pricing
7
may be too low to generate an acceptable return. We are not seeking regulatory approval to market Korlym outside the United States.
Coverage and Reimbursement
Sales of our products will depend, in part, on the extent to which they will be covered by government health care programs and commercial insurance and managed healthcare organizations. Third-party payers are increasingly limiting coverage and reducing reimbursements for medical products and services, although this trend has not to-date had a material impact on the amount or timing of our revenues. In addition, the U.S. government, state legislatures and foreign governments have continued implementing cost-containment programs, including price controls, restrictions on coverage and reimbursement and requirements for substitution of generic products. Adoption of price controls and cost-containment measures and adoption of more restrictive policies in jurisdictions with existing controls and measures could limit our revenue. Decreases in third-party reimbursement for our products or a decision by a third-party payer to not cover our products could reduce our sales and have a material adverse effect on our results of operations and financial condition.
Other Healthcare Laws
We are subject to healthcare regulation and enforcement by the federal government and the states in which we conduct our business. These laws include, without limitation, state and federal anti-kickback, fraud and abuse, false claims, privacy and security and physicians’ sunshine laws and regulations. Foreign governments have comparable regulations.
The federal Anti-Kickback Statute prohibits, among other things, any person from knowingly and willfully offering, soliciting, receiving or providing remuneration, directly or indirectly, to induce either the referral of an individual, for an item or service or the purchasing or ordering of a good or service, for which payment may be made under federal healthcare programs such as the Medicare and Medicaid programs. The Anti-Kickback Statute is subject to evolving interpretations. In the past, the government has enforced the Anti-Kickback Statute to reach large settlements with healthcare companies based on sham consulting and other financial arrangements with physicians. Further, a person or entity does not need to have actual knowledge of these statutes or specific intent to violate them to have committed a violation. The majority of states also have anti-kickback laws which establish similar prohibitions and in some cases may apply to items or services reimbursed by any third-party payor, including commercial insurers.
Additionally, the civil False Claims Act prohibits knowingly presenting or causing the presentation of a false, fictitious or fraudulent claim for payment to the U.S. government. Actions under the False Claims Act may be brought by the Attorney General or as a qui tam action by a private individual in the name of the government. In addition, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal False Claims Act. Violations of the False Claims Act can result in very significant monetary penalties and treble damages. The federal government is using the False Claims Act, and the accompanying threat of significant liability, in its investigation and prosecution of pharmaceutical and biotechnology companies in connection with the promotion of products for unapproved uses and other sales and marketing practices. The government has obtained multi-billion dollar settlements under the False Claims Act in addition to individual criminal convictions under applicable criminal statutes. We expect that the government will continue to devote substantial resources to investigating healthcare providers’ and manufacturers’ compliance with applicable fraud and abuse laws.
The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, prohibits, among other things, knowingly and willfully executing a scheme to defraud any healthcare benefit program. In addition, HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, and their implementing regulations, imposes certain requirements relating to the privacy, security and transmission of protected health information on HIPAA covered entities, which include certain healthcare providers, health plans and healthcare clearinghouses, and their business associates who conduct certain activities for or on their behalf involving protected health information on their behalf. Even when HIPAA does not apply, according to the Federal Trade Commission or the FTC, failing to take appropriate steps to keep consumers’ personal information secure constitutes unfair acts or practices in or affecting commerce in violation of Section 5(a) of the Federal Trade Commission Act, or the FTCA, 15 U.S.C § 45(a). The FTC expects a company’s data security measures to be reasonable and appropriate in light of the sensitivity and volume of consumer information it holds, the size and complexity of its business, and the cost of available tools to improve security and reduce vulnerabilities. Individually identifiable health information is considered sensitive data that merits stronger safeguards. The FTC’s guidance for appropriately securing consumers’ personal information is similar to what is required by the HIPAA Security Rule.
In addition, there has been increased federal and state regulation of payments made to physicians and other healthcare providers. The Patent Protection and Affordable Care Act ("PPACA"), among other things, imposes new reporting requirements on drug manufacturers for payments made by them to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors), certain health care professionals beginning in 2022 and teaching hospitals, as well as ownership and investment
8
interests held by physicians and their immediate family members. Failure to submit required information may result in significant civil monetary penalties for any payments, transfers of value or ownership or investment interests that are not timely, accurately and completely reported in an annual submission. Drug manufacturers must report such payments to the government by the 90th day of each calendar year. Certain states also mandate implementation of commercial compliance programs, impose restrictions on drug manufacturer marketing practices and/or require the tracking and reporting of gifts, compensation and other remuneration to physicians.
State and foreign laws and regulations restrict business practices in the pharmaceutical industry and complicate our compliance efforts. For example, some states require companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the federal government’s compliance guidance or otherwise restrict payments to healthcare providers and other potential referral sources. Some states require manufacturers to file reports relating to pricing and marketing information. Some state and local governments require the public registration of pharmaceutical sales representatives.
Certain state and foreign laws also govern the privacy and security of health information in ways that differ significantly from one another and are not preempted by HIPAA. For example, California recently enacted legislation, the California Consumer Privacy Act, or CCPA, which went into effect January 1, 2020. The CCPA, among other things, creates new data privacy obligations for covered companies and provides new privacy rights to California residents, including the right to opt out of certain disclosures of their information. The CCPA also creates a private right of action with statutory damages for certain data breaches, thereby potentially increasing risks associated with a data breach. Although the law includes limited exceptions, including for “protected health information” maintained by a covered entity or business associate, it may regulate or impact our processing of personal information depending on the context. In Europe, the General Data Protection Regulation, or GDPR, went into effect in May 2018 and imposes stringent data protection requirements for controllers and processors of personal data of persons within the EU. The GDPR applies to any company established in the EU as well as to those outside the EU if they collect and use personal data in connection with the offering of goods or services to individuals in the EU or the monitoring of their behavior. Companies that must comply with the GDPR face increased compliance obligations and risk, including more robust regulatory enforcement of data protection requirements and potential fines for noncompliance of up to €20 million or 4% of the annual global revenues of the noncompliant company, whichever is greater. In addition, the United Kingdom leaving the EU could also lead to further legislative and regulatory changes. It remains unclear how the United Kingdom data protection laws or regulations will develop in the medium to longer term and how data transfer to the United Kingdom from the EU will be regulated, especially following the United Kingdom's departure from the EU on January 31, 2020 without a deal. However, the United Kingdom has transposed the GDPR into domestic law with the Data Protection Act 2018, which remains in force following the United Kingdom's departure from the EU.
The shifting commercial compliance environment and the need to build and maintain robust and expandable systems to comply with different compliance and/or reporting requirements in multiple jurisdictions increase the possibility that a healthcare company may violate one or more of the requirements. If our operations are found to be in violation of any of such laws or any other governmental regulations that apply to us, we may be subject to penalties, including, without limitation, civil and criminal penalties, damages, fines, the curtailment or restructuring of our operations, exclusion from participation in federal and state healthcare programs and imprisonment, any of which could adversely affect our ability to operate our business and our financial results.
Employees
We are managed by experienced pharmaceutical executives. We also enlist the expertise of advisors with extensive pharmaceutical experience. As of December 31, 2019, we had 206 employees, five of whom have MDs. We consider our employee relations to be good. Our employees are not covered by a collective bargaining agreement.
About Corcept
We were incorporated in the State of Delaware on May 13, 1998. Our registered trademarks include Corcept® and Korlym®. Other service marks, trademarks and trade names referred to in this document are the property of their respective owners.
Available Information
We are subject to the information requirements of the Securities Exchange Act of 1934, as amended, and we therefore file periodic reports, proxy statements and other information with the SEC relating to our business, consolidated financial statements and other matters. The SEC maintains an Internet site, www.sec.gov, that contains reports, proxy statements and other information regarding issuers such as Corcept.
For more information about Corcept, including free access to our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports, visit our website at www.corcept.com or the SEC’s website,
9
www.sec.gov. The information found on or accessible through our website is not incorporated into, and does not form a part of, this Form 10-K.
ITEM 1A. RISK FACTORS
Investing in our common stock involves significant risks. Before investing, carefully consider the risks described below and the other information in this Annual Report on Form 10-K, including our consolidated financial statements and related notes. The risks and uncertainties described below are the ones we believe may materially affect us. However, there may be others of which we are unaware that could materially harm our business or financial condition and cause the price of our stock to decline, in which case you could lose all or part of your investment.
Risks Related to our Commercial Activities
Failure to generate sufficient revenue from the sale of Korlym would harm our financial results and would likely cause our stock price to decline.
Our ability to generate revenue and to fund our commercial operations and development programs is dependent on the sale of Korlym to treat patients with Cushing’s syndrome. Physicians will prescribe Korlym only if they determine that it is preferable to other treatments, even if those treatments are not approved for Cushing’s syndrome. Because Cushing’s syndrome is rare, most physicians are inexperienced diagnosing or caring for patients with the illness and it can be hard to persuade them to identify appropriate patients and treat them with Korlym.
Many factors could limit our Korlym revenue, including:
• | the preference of some physicians for off-label treatments for Cushing’s syndrome, such as ketoconazole; |
• | competition from non-medical treatments, such as surgery and radiation; |
• | the potential introduction of a competitor for Korlym, including a generic version of Korlym; |
• | the lack of availability of adequate private and government insurance coverage; |
• | negative publicity and political concerns about Korlym’s active ingredient, mifepristone, which is approved in another drug for the termination of pregnancy; and |
• | technological change that makes Korlym obsolete. |
Failure to generate sufficient Korlym revenue may prevent us from fully funding our planned commercial and clinical activities and would likely cause our stock price to decline.
If generic versions of Korlym are approved and successfully commercialized, our business, results of operations and financial position would be adversely affected.
The marketing exclusivity provided by Korlym’s orphan drug designation expired in February 2019. Other companies may now seek to introduce generic equivalents of Korlym for the treatment of Cushing’s syndrome, provided they receive FDA approval and can show that their products do not infringe patents we hold covering Korlym’s use to treat patients with Cushing’s syndrome or that these patents are invalid or unenforceable. If our patents are successfully challenged and a generic version of Korlym becomes available, our sales of Korlym tablets and their price could decline rapidly and significantly, which would reduce our revenue and materially harm our results of operations and financial position. Competition from a generic version of Korlym may also cause our revenue to be materially less than the public guidance we have provided, which would likely cause the price of our common stock to decline.
We have sued Teva and Sun in Federal District Court with respect to their proposed generic versions of Korlym. Litigation to enforce or defend intellectual property rights is complex, costly and involves significant commitments of management time. There can be no assurance of a successful outcome. Please see “Part I, Item 3, Legal Proceedings.” Furthermore, on August 1, 2020, after the 30-month stay provided by the Hatch-Waxman Act has expired, Teva may choose to market a generic version of Korlym, notwithstanding any ongoing litigation with us. Even if we prevail in our legal action and Teva withdraws its product and pays us damages, the temporary availability of a generic version of Korlym could materially harm our results of operations and financial condition.
10
Other companies offer or are attempting to develop different medications to treat patients with Cushing’s syndrome. The availability of competing treatments could limit our revenue from Korlym.
Since 2012, a medication developed by Novartis and now owned by the Italian pharmaceutical company Recordati, the somatostatin analogue Signifor® (pasireotide) Injection, has been marketed in both the United States and the EU for adult patients with Cushing’s disease (a subset of Cushing’s syndrome). Recordati is also developing the cortisol synthesis inhibitor osilodrostat to treat patients with Cushing’s syndrome. Osilodrostat has been designated an orphan drug for that indication in both the EU and the United States. The EU’s Committee for Medicinal Products for Human Use recommended that osilodrostat be approved in the EU for the treatment of endogenous Cushing’s syndrome. Recordati has announced that it also plans to seek marketing approval for osilodrostat in the United States.
Strongbridge Biopharma plc (“Strongbridge”) has received orphan drug designation in the United States and the EU for the use of the cortisol synthesis inhibitor levoketoconazole to treat patients with Cushing’s syndrome. Levoketoconazole is an enantiomer of the generic anti-fungal medication, ketoconazole, that is prescribed off-label to treat patients with Cushing’s syndrome. Strongbridge has completed one Phase 3 trial, which met its primary endpoint of reducing cortisol synthesis, and is conducting a second Phase 3 trial.
If we cannot continue to obtain acceptable prices or adequate insurance coverage and reimbursement for Korlym, we will be unable to generate significant revenues.
The commercial success of Korlym depends on the availability of adequate insurance coverage and reimbursement. Government payers, including Medicare, Medicaid and the Veterans Administration, as well as private insurers and health maintenance organizations, are increasingly attempting to contain healthcare costs by limiting reimbursement for medicines. If government or private payers cease to provide adequate and timely coverage and reimbursement for Korlym, physicians may not prescribe the medication and patients may not purchase it, even if it is prescribed. In addition, delays in coverage for individual patients may reduce our revenues.
In some foreign markets, drug prices and the profitability of prescription medications are subject to government control. In the United States, we expect that there will continue to be federal and state proposals for similar controls. Also, the trends toward managed health care in the United States and recent laws and legislation intended to increase the public visibility of drug prices and reduce the cost of government and private insurance programs could significantly influence the purchase of health care services and products and may result in lower prices for Korlym.
In the United States, there have been and continue to be a number of legislative initiatives to contain healthcare costs. The Patient Protection and Affordable Care Act (“PPACA”), which was passed in 2010, substantially changed the way health care is financed by both governmental and private insurers. The PPACA, among other things, expanded Medicaid program eligibility and access to commercial health insurance coverage, increased the minimum Medicaid rebates owed by manufacturers under the Medicaid Drug Rebate Program and extended the rebate program to individuals enrolled in Medicaid managed care organizations, established annual fees and taxes on manufacturers of certain branded prescription drugs, and promoted a new Medicare Part D coverage gap discount program. The PPACA also appropriated funding to comparative clinical effectiveness research, although it remains unclear how the research will affect Medicare coverage and reimbursement or how new information will influence other third-party payer policies.
Since its enactment, there have been judicial and Congressional challenges to certain aspects of the PPACA, and we expect there will be additional challenges and amendments to the PPACA in the future. For example, the Tax Cuts and Jobs Acts (the “Tax Act”) was enacted, which, among other things, removed penalties for not complying with the individual mandate to carry health insurance. On December 14, 2018, a U.S. District Court Judge in the Northern District of Texas, ruled that the individual mandate is a critical and inseverable feature of the PPACA, and therefore, because it was repealed as part of the Tax Act, the remaining provisions of the PPACA are invalid as well. On December 18, 2019, the U.S. Court of Appeals for the 5th Circuit upheld the district court’s decision that the individual mandate was unconstitutional but remanded the case back to the District Court to determine whether the remaining provisions of the Affordable Care Act are invalid as well. It is unclear how these decisions, subsequent appeals, if any, and other efforts to challenge, replace or repeal the PPACA will affect the law or our business. Any new limitations on, changes to, or uncertainty with respect to the ability of individuals to enroll in governmental reimbursement programs or other third-party payer insurance plans could reduce Korlym sales, which in turn could affect our ability to successfully develop and commercialize new products.
Other legislative and regulatory changes have been proposed and adopted in the United States since the PPACA was enacted. These changes included an aggregate reduction in Medicare payments to providers of 2 percent per fiscal year, which went into effect on April 1, 2013 and will remain in effect through 2029 unless additional Congressional action is taken, and the American Taxpayer Relief Act of 2012, which further reduced Medicare payments to several providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from
11
three to five years. Moreover, the federal government and the individual states in the United States have become increasingly active in developing proposals, passing legislation and implementing regulations designed to control drug pricing, including price or patient reimbursement constraints, discounts, formulary flexibility, marketing cost disclosure and transparency measures.
These new laws and the regulations and policies implementing them, as well as other healthcare-related measures that may be adopted in the future, could materially reduce our ability to develop and commercialize our product candidates.
The unfavorable public perception of mifepristone may limit our ability to sell Korlym.
The active ingredient in Korlym, mifepristone, is approved by the FDA in another drug for the termination of early pregnancy. As a result, mifepristone is the subject of considerable debate in the United States and elsewhere. Public perception of mifepristone may limit the acceptance of Korlym by patients and physicians. Even though we have taken measures to minimize the chance that Korlym will accidentally be prescribed to a pregnant woman, physicians may choose not to prescribe Korlym to a woman simply to avoid the risk of terminating a pregnancy.
We depend on vendors to manufacture Korlym’s active ingredient, form it into tablets, package it and dispense it to patients. We also depend on vendors to manufacture the API and capsules or tablets for our product candidates. If our suppliers become unable or unwilling to perform these functions and we cannot transfer these activities to replacement vendors in a timely manner, our business will be harmed.
A single third-party manufacturer, PCAS, supplies the API in Korlym. Two other third-party manufacturers produce and bottle Korlym tablets. Our agreement with PCAS automatically renews for two one-year terms, unless either party provides 12-months’ written notice of its intent not to renew. A single specialty pharmacy, Optime Care, Inc. (“Optime”) dispenses the Korlym we sell directly to patients and collects payments from insurers and other payers representing approximately 99 percent of our revenue. If Optime does not adhere to its agreements with payers, it may not be able to collect some or all of the payments due to us. Our agreement with Optime has a five-year term and renews upon the written consent of both parties, subject to customary termination provisions. In addition, we may terminate the agreement for convenience.
The facilities used by our vendors to manufacture and package the API and drug product of Korlym and our product candidates must be approved by the FDA and, in some cases, the European Medicines Agency (“EMA”). We do not control the activities of these vendors, including whether they maintain adequate quality control and hire qualified personnel. We are dependent on them for compliance with the regulatory requirements known as current good manufacturing practices (“cGMPs”). If our vendors cannot manufacture material that conforms to our specifications and the strict requirements of the FDA or others, they will not be able to maintain regulatory authorizations for their facilities and we could be prohibited from using the API or drug product they have provided. If the FDA, EMA or other regulatory authorities withdraw regulatory authorizations of these facilities, we may need to find alternative vendors or facilities, which would be time-consuming, complex and expensive and could significantly hamper our ability to develop, obtain regulatory approval for and market our products. Sanctions could be imposed on us, including fines, injunctions, civil penalties, refusal of regulators to approve our product candidates, delays, suspensions or withdrawals of approvals, seizures or recalls of products, operating restrictions and criminal prosecutions, any of which could harm our business.
We may not have adequate insurance to cover our exposure to product liability claims.
We may be subject to product liability or other claims based on allegations that Korlym or one of our product candidates has harmed a patient. Such a claim may damage our reputation by raising questions about Korlym or our product candidates’ safety and could prevent or interfere with product development or commercialization. Less common adverse effects of a pharmaceutical product are sometimes not known until long after the product is approved for marketing. Because the active ingredient in Korlym is used to terminate pregnancy, clinicians using Korlym in clinical trials and physicians prescribing the medicine to women must take strict precautions to ensure that it is not administered to pregnant women. Failure to observe these precautions could result in significant product liability claims.
Our product liability insurance may not fully cover our liabilities. Inability to obtain adequate insurance coverage could inhibit development of our product candidates or result in significant uninsured liability. Defending a lawsuit could be costly and divert management from productive activities.
If we are unable to maintain regulatory approval of Korlym for the treatment of patients with Cushing’s syndrome or if we fail to comply with other requirements, we will be unable to generate revenue and may be subject to penalties.
We are subject to oversight by the FDA and other regulatory authorities in the United States and elsewhere with respect to our research, testing, manufacturing, labeling, distribution, adverse event reporting, storage, advertising, promotion, recordkeeping and sales and marketing activities. These requirements include submissions of safety information, annual updates on manufacturing activities and continued compliance with FDA regulations, including cGMPs, good laboratory practices and good clinical practices
12
(“GCP”). The FDA enforces these regulations through inspections of us and the laboratories, manufacturers and clinical sites we use. Foreign regulatory authorities have comparable requirements and enforcement mechanisms. Discovery of previously unknown problems with a product or product candidate, such as adverse events of unanticipated severity or frequency or deficiencies in manufacturing processes or management, as well as failure to comply with FDA or other U.S. or foreign regulatory requirements, may subject us to substantial civil and criminal penalties, injunctions, holds on clinical trials, product seizure, refusal to permit the import or export of products, restrictions on product marketing, withdrawal of the product from the market, product recalls, total or partial suspension of production, refusal to approve pending NDAs or supplemental NDAs, and suspension or revocation of product approvals.
We cannot predict how government regulations may change. The Trump administration has taken actions that could impose significant burdens on or materially delay the FDA’s ability to implement new rules, issue guidance and review and approve marketing applications. It is difficult to predict how these executive actions will be implemented, if at all, and the extent to which they will affect the FDA’s ability to exercise its authority. If these executive actions impair the FDA’s ability to carry out its regulatory responsibilities or if we are slow or unable to adapt to sudden changes in regulatory requirements, our regulatory compliance may lapse and we may lose marketing approval for Korlym or face enforcement action.
We may be subject to civil or criminal penalties if our marketing of Korlym violates FDA regulations or health care fraud and abuse laws.
We are subject to FDA regulations governing the promotion and sale of medications. Although physicians are permitted to prescribe drugs for any indication they choose, manufacturers may only promote products for their FDA-approved use. All other uses are referred to as “off-label.” In the United States, we market Korlym to treat hyperglycemia secondary to hypercortisolism in adult patients with endogenous Cushing’s syndrome who have type 2 diabetes mellitus or glucose intolerance and for whom surgery has failed or is not an option. We provide promotional materials and training programs to physicians covering the use of Korlym for this indication. The FDA may change its policies or enact new regulations at any time that restrict our ability to promote our products.
Although we believe our marketing materials and training programs do not constitute “off-label” promotion, the FDA may disagree. If the FDA determines that our promotional materials, training or other activities by our employees or agents constitute “off-label” promotion, it could require us to change them. The FDA could also subject us to regulatory enforcement actions, including issuance of a public “warning letter,” injunction, seizure, civil fine or criminal penalties. Other federal or state enforcement authorities might act if they believe that the alleged improper promotion led to the submission and payment of claims for an unapproved use, which could result in significant fines or penalties under other statutory authorities, such as laws prohibiting false claims for reimbursement. Even if it is determined that we are not in violation of these laws, we may receive negative publicity, incur significant expenses and be forced to devote management time to defending our position.
We are subject to federal and state healthcare fraud and abuse regulations, including:
• | the federal Anti-Kickback Statute, which prohibits, among other things, knowingly and willfully soliciting, receiving, offering or paying remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual for, or the purchase, order or recommendation of, any good or service for which payment may be made under federal health care programs such as Medicare and Medicaid. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation; |
• | federal false claims laws, including, without limitation, the False Claims Act, which prohibit any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government, or knowingly making, or causing to be made, a false statement to get a false claim paid. In addition, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act. Pharmaceutical companies have been prosecuted under these laws for a variety of promotional and marketing activities, such as allegedly providing free product to or entering into “sham” consulting arrangements with customers to induce such customers to purchase, order or recommend the company’s products in violation of the Anti-Kickback Statute and federal false claims laws and regulations; reporting to pricing services inflated average wholesale prices that were then used by certain governmental programs to set reimbursement rates; engaging in the promotion of “off-label” uses that caused customers to submit claims to and obtain reimbursement from governmental payers for non-covered “off-label” uses; and submitting inflated best price information to the Medicaid Drug Rebate Program; the government may assert that a claim including items and services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act; |
• | the federal Civil Monetary Penalties law, which prohibits, among other things, offering or transferring remuneration to a federal healthcare beneficiary that a person knows or should know is likely to influence the beneficiary’s decision to order or receive items or services reimbursable by the government from a particular provider or supplier; |
13
• | the federal Health Insurance Portability and Accountability Act of 1996 (“HIPAA”), which created federal criminal laws that prohibit executing a scheme to defraud any health care benefit program or making false statements relating to health care matters; |
• | federal “sunshine” laws, including the federal Physician Payment Sunshine Act, that require transparency regarding financial arrangements with health care providers, such as the reporting and disclosure requirements imposed by the PPACA on drug manufacturers regarding any “transfer of value” made or distributed to physicians (defined to include doctors, dentists, optometrists, podiatrists and chiropractors), certain health care professionals beginning in 2022, teaching hospitals, and ownership or investment interests held by physicians and their immediate family members. Manufacturers are required to submit reports detailing these financial arrangements by the 90th day of each calendar year; |
• | federal consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers; and |
• | state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws which may apply to items or services reimbursed by any third-party payer, including commercial insurers; state laws that require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government or otherwise restrict payments that may be made to healthcare providers; and state laws that require drug manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures and pricing information. |
The risk of our operations being found in violation of these laws and regulations is increased by the fact that many of them have not been definitively interpreted by regulatory authorities or the courts and their provisions are open to a variety of interpretations. Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available under them, it is possible that some of our business activities, including our relationships with physicians and other healthcare providers (some of whom recommend, purchase and/or prescribe our products) and the manner in which we promote our products, could be subject to challenge. We are also exposed to the risk that our employees, independent contractors, principal investigators, consultants, vendors, distributors, and contract research organizations (“CROs”) may engage in fraudulent or other illegal activity. Although we have policies and procedures prohibiting such activity, it is not always possible to identify and deter misconduct and the precautions we take may not be effective in controlling unknown risks or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with applicable laws and regulations.
If our operations are found to be in violation of any of the laws described above or any other government regulations, we may be subject to civil and criminal penalties, damages, fines, exclusion from governmental health care programs, a corporate integrity agreement or other agreement to resolve allegations of non-compliance, individual imprisonment, and the curtailment or restructuring of our operations, any of which could adversely affect our financial results and ability to operate.
A breakdown or breach of our information technology systems or our failure to protect confidential information concerning patients or others could subject us to liability or interrupt the operation of our business.
We store valuable confidential information relating to our business, patients and employees on our computer networks and on the networks of our vendors. Despite the implementation of security measures, these networks are subject to the risk of cyberattacks, computer viruses, unauthorized access, natural disasters, terrorism, war and internet and electrical failures. They may also be manipulated by criminals seeking to commit fraud or theft. In addition, system failures could cause the loss or theft of valuable clinical trial data or otherwise disrupt our clinical and commercial activities and be expensive and time-consuming to remedy. If a disruption or security breach resulted in the disclosure of confidential or proprietary information, we could incur liability and our research, development and commercialization efforts could be delayed or otherwise harmed.
We are subject to government regulation and other legal obligations relating to privacy and data protection. Compliance with these requirements is complex and costly. Failure to comply could materially harm our business.
We are subject to statutes concerning data privacy and security, including HIPAA and the EU’s General Data Protection Regulation (“GDPR”). These and other regulatory frameworks are evolving rapidly as new rules are enacted and existing ones updated and made more stringent.
In the United States, HIPAA imposes privacy, security and breach reporting obligations with respect to individually identifiable health information upon “covered entities” (health plans, health care clearinghouses and certain health care providers), and their respective business associates, individuals or entities that create, received, maintain or transmit protected health information in connection with providing a service for or on behalf of a covered entity. HIPAA mandates the reporting of certain breaches of health information to HHS, affected individuals and if the breach is large enough, the media. Entities that are found to be in violation of HIPAA as the result of a breach of unsecured protected health information, a complaint about privacy practices or an
14
audit by HHS, may be subject to significant civil, criminal and administrative fines and penalties and/or additional reporting and oversight obligations if required to enter into a resolution agreement and corrective action plan with HHS to settle allegations of HIPAA non-compliance. Even when HIPAA does not apply, according to the Federal Trade Commission or the FTC, failing to take appropriate steps to keep consumers’ personal information secure constitutes unfair acts or practices in or affecting commerce in violation of Section 5(a) of the Federal Trade Commission Act, or the FTCA, 15 U.S.C § 45(a). The FTC expects a company’s data security measures to be reasonable and appropriate in light of the sensitivity and volume of consumer information it holds, the size and complexity of its business, and the cost of available tools to improve security and reduce vulnerabilities. Individually identifiable health information is considered sensitive data that merits stronger safeguards. The FTC’s guidance for appropriately securing consumers’ personal information is similar to what is required by the HIPAA Security Rule.
In addition, certain state laws govern the privacy and security of health information in certain circumstances, some of which are more stringent than HIPAA and many of which differ from each other in significant ways and may not have the same effect, thus complicating compliance efforts. Failure to comply with these laws, where applicable, can result in the imposition of significant civil and/or criminal penalties and private litigation. For instance, on June 28, 2018, California enacted the California Consumer Privacy Act, or CCPA, which took effect on January 1, 2020. The CCPA creates individual privacy rights for California consumers and increases the privacy and security obligations of entities handling certain personal information. The CCPA provides for civil penalties for violations, as well as a private right of action for data breaches that is expected to increase data breach litigation. The CCPA may increase our compliance costs and potential liability. Similar laws have been proposed at the federal level and in other states.
The GDPR took effect in 2018. It establishes new requirements for the use and safeguarding of personal data in the EU and applies to companies established in the EU as well as companies that collect and use personal data to offer goods or services to, or monitor the behavior of, individuals in the EU (including in clinical trials). Penalties for failure to comply include fines of up to €20 million or four percent of worldwide annual revenue, whichever is greater. Data protection authorities in some of the EU member states have not completed their interpretative guidance and implementing laws and regulations, which makes compliance with the GDPR difficult. In addition, data protection authorities of the different EU countries may interpret GDPR requirements differently. Once promulgated, national and EU guidance will likely be updated from time to time, which will add complexity and cost to our collection and handling of data. In addition, the United Kingdom leaving the EU could also lead to further legislative and regulatory changes. It remains unclear how the United Kingdom data protection laws or regulations will develop in the medium to longer term and how data transfer to the United Kingdom from the EU will be regulated, especially following the United Kingdom's departure from the EU on January 31, 2020 without a deal. However, the United Kingdom has transposed the GDPR into domestic law with the Data Protection Act 2018, which remains in force following the United Kingdom's departure from the EU.
Complying with HIPAA, the GDPR and other data privacy and security requirements is complex and costly. Failure to comply by us or our vendors could subject us to litigation, government enforcement actions and substantial penalties and fines, which could harm our business.
We are dependent on the continued functioning of the FDA and other federal instrumentalities. Inadequate funding of these instrumentalities, their partial or complete closure, or their inability to hire and retain talented professionals due to uncertainties about their ability to pay their employees could materially harm our business.
The FDA’s ability to carry out its mandated functions is affected by a variety of factors, including adequate government funding, the ability to hire and retain key personnel, and statutory, regulatory and policy changes. Disruptions at the FDA and other agencies may slow the time to review new drug applications and respond to other inquiries. Disruptions at the Securities and Exchange Commission (“SEC”) may temporarily stop its ability to review and approve proposed financing transactions. Several times in the last few years, including for 35 days beginning on December 22, 2018, the U.S. government has shut down and many regulatory agencies, including the FDA and SEC, have had to furlough employees and stop critical activities. If a prolonged government shutdown occurs, it could significantly impair the FDA, SEC and other authorities' ability to process our submissions, which could materially harm our business.
In addition, many of our patients pay for Korlym with insurance or other support provided by payers who are funded in whole or in part by the U.S. federal government, such as Medicare, Medicaid, Tricare and the Veterans Administration. If a partial or total shutdown of the federal government prevents these payers from funding their obligations, our revenues could decline.
Changes in federal, state and local tax laws may reduce our net earnings.
Our earnings are subject to federal, state and local tax. We offset a portion of our earnings using net operating losses and our taxes using research and development tax credits, which reduces the amount of tax we pay. Some jurisdictions require that we pay taxes or fees calculated as a percentage of sales, payroll expense, or other indicia of our activities. Please see “Part IV, Item 16, Notes to Consolidated Financial Statements - Income Taxes.” Changes to existing tax laws that we cannot control or predict could materially increase the amount of taxes and fees we must pay. For example, an increase in income tax rates or a reduction or
15
elimination of net operating losses and research and development tax credits could significantly increase our tax expense, which would reduce our net income and adversely affecting our results of operations.
A disaster could damage our own or our manufacturers’ facilities and equipment, which could require us to cease or curtail operations.
Our business is vulnerable to damage from various types of natural disasters or other disruptive events, including earthquakes, fires, floods, power losses and communications failures. Our headquarters are in the San Francisco Bay Area, which is earthquake-prone. Our specialty pharmacy and tablet manufacturer are in areas subject to hurricanes and tornadoes. Political considerations relating to mifepristone put us and our manufacturers at increased risk of protests and disruptive events. If a disaster were to occur, we might not be able to operate our business. Our insurance may not cover or be adequate to cover losses resulting from disasters or other business interruptions.
Risks Related to our Research and Development Activities
Clinical drug development is lengthy, expensive and often unsuccessful. Results of early studies and trials are often not predictive of later trial results. Failure can occur at any stage of drug development. Our efforts to discover, develop and commercialize our product candidates may not succeed.
Clinical development is expensive, lengthy and often unsuccessful. Data from clinical trials are susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. The results from early clinical trials are often not predictive of results in later clinical trials. Product candidates may fail to show the desired safety and efficacy traits despite having produced positive results in preclinical studies and initial clinical trials. Many companies have suffered significant setbacks in late-stage clinical trials due to lack of efficacy or unanticipated or unexpectedly severe adverse events.
Our current clinical trials may prove inadequate to support marketing approvals. Even trials that generate positive results may have to be confirmed in much larger, more expensive and lengthier trials before we could realistically seek regulatory approval of a product candidate.
Clinical trials may be delayed by many factors, including:
• | delays obtaining regulatory permission to start a trial, changes to the size or design of a trial or changes in regulatory requirements for a trial already underway; |
• | inability to secure acceptable terms with vendors and an appropriate number of clinical trial sites; |
• | delays or inability to obtain institutional review board (“IRB”) approval at prospective trial sites; |
• | slow patient enrollment; |
• | failure of patients or investigators to comply with the clinical trial protocol; |
• | unforeseen safety issues; and |
• | negative findings of inspections of clinical sites or manufacturing operations by us, the FDA or other authorities. |
A trial may be suspended or terminated by us, the trial’s data safety monitoring board, the IRBs governing the sites where the trial is being conducted or the FDA for many reasons, including failure to comply with regulatory requirements or clinical protocols, negative findings in an inspection of our clinical trial operations or trial sites by the FDA or other authorities, unforeseen safety issues, failure to demonstrate a benefit or changes in government regulations.
During the development of a product candidate, we may decide, or the FDA or other regulatory authorities may require us, to conduct more pre-clinical or clinical studies or to change the size or design of a trial already underway, which could delay or prevent the completion of development and increase its cost. Even if we conduct all of the clinical trials and supportive studies that we consider appropriate and the results are positive, we may not receive regulatory approval.
Vendors manufacture and distribute the drug product we use in our trials, conduct and manage some of our clinical trials and perform data collection and analysis. Failure of these vendors to perform their duties or meet expected timelines may prevent or delay approval of our product candidates.
Third-party clinical investigators and clinical sites enroll patients and CROs manage many of our trials and perform data collection and analysis. Although we control only certain aspects of these third-parties’ activities, we are responsible for ensuring that every study adheres to its protocol and meets regulatory and scientific standards. If any of our vendors does not perform its duties or
16
meet expected deadlines or fails to adhere to applicable GCP, or if the quality or accuracy of the data it produces is compromised, affected clinical trials may be extended, delayed or terminated and we may be unable to obtain approval for our product candidates. Failure of our manufacturing vendors to perform their duties or comply with cGMPs may require us to recall drug product or repeat clinical trials, which would delay regulatory approval. If our agreements with any of these vendors terminate, we may not be able to enter into alternative arrangements in a timely manner or on reasonable terms.
We may be unable to obtain or maintain regulatory approvals for our product or product candidates.
We cannot promote a product candidate unless the FDA or comparable foreign regulatory authorities approves it, which may not happen. Obtaining regulatory approval of a drug is difficult, uncertain, lengthy and expensive. Failure can occur at any stage. In order to receive FDA approval, we must demonstrate to the FDA's satisfaction that the new drug is safe and effective for its intended use and that our manufacturing processes comply with cGMPs. Our inability or the inability of our vendors to comply with applicable FDA and other regulatory requirements can result in delays in or denials of new product approvals, warning letters, fines, consent decrees restricting or suspending manufacturing operations, injunctions, civil penalties, recall or seizure of products, total or partial suspension of product sales and criminal prosecution. Any of these or other regulatory actions could materially harm our business and financial condition.
If we receive regulatory approval for a product candidate, we will be subject to ongoing FDA requirements and oversight, such as continued safety and other reporting requirements and post-marketing restrictions. If we are not able to maintain regulatory compliance, we may not be permitted to develop our product candidates or market our products and may be subject to product recalls or seizures. Any regulatory approvals for our product candidates may require costly post-marketing studies. Future governmental action or changes in FDA policy or personnel may also result in delays or rejection of an NDA or supplemental NDA.
Obtaining regulatory approval of product candidates in foreign jurisdictions would be costly and difficult. Failure to obtain such approvals would prevent us from commercializing our product candidates outside the United States.
We may seek to commercialize our products in international markets, which would require us to receive a marketing authorization and, in many cases, pricing approval, from the appropriate regulatory authorities. These approval processes include all of the risks associated with the FDA’s approval process and, in some cases, more. Approval procedures vary between countries and can require additional pre-clinical or clinical studies. Obtaining approval may take longer than it does in the United States. Although approval by the FDA does not ensure approval by regulatory authorities in other countries, and approval by one foreign regulatory authority does not ensure approval by others, failure or delay in obtaining regulatory approval in one country may have a negative effect on the regulatory process in others.
Our products and product candidates may cause undesirable side effects that halt their clinical development, prevent their regulatory approval, limit their commercial potential or cause us significant liability.
Patients in clinical trials report changes in their health, including new illnesses, injuries, and discomforts, to their study doctor. Often, it is not possible to determine whether or not these conditions were caused by the drug candidate being studied or something else. As we test our product candidates in larger, longer and more extensive clinical trials, or as use of them becomes more widespread if receive regulatory approval, patients may report serious adverse events that did not occur or went undetected in previous trials. Many times, serious side effects are only detected in large-scale, Phase 3 clinical trials or following commercial approval.
Adverse events reported in clinical trials can slow or stop patient recruitment, prevent enrolled patients from completing a trial and could give rise to liability claims. Regulatory authorities could respond to reported adverse events by interrupting or halting our clinical trials or limiting the scope of, delaying or denying marketing approval. If we elect, or are required by authorities, to delay, suspend or terminate any clinical trial or commercialization efforts, the commercial prospects of such product candidates or products may be harmed, and our ability to generate product revenues from them may be delayed or eliminated.
If one of our product candidates receives marketing approval, and we or others later identify undesirable side effects or adverse events, potentially significant negative consequences could result, including but not limited to:
• | regulatory authorities may suspend, limit or withdraw approvals of such product; |
• | regulatory authorities may require additional warnings on the label, including “boxed” warnings, or issue safety alerts other safety information about the product; |
• | we may be required to change the way the product is administered or conduct additional studies or clinical trials; |
17
• | we may be required to create a Risk Evaluation and Mitigation Strategy (REMS), which could include a medication guide outlining the risks of such side effects for distribution to patients, a communication plan for healthcare providers and/or other elements to assure safe use; |
• | the product may become less competitive; |
• | we may be subject to fines, injunctions or the imposition of criminal penalties; and |
• | we could be sued and held liable for harm caused to patients; |
Any of these events could seriously harm our business.
We may face competition from companies with greater financial, technical and marketing resources than our own.
The pharmaceutical industry is competitive and subject to rapid technological change. Our potential competitors include large pharmaceutical companies, which have greater resources than our own and may develop and commercialize medications that are superior to and less expensive than ours, which could negatively affect our financial results.
We need to increase the size of our organization and may experience difficulties in managing growth.
Our commercial and research and development efforts are constrained by our limited administrative, operational and management resources. To date, we have relied on a small management team. Growth will impose significant added responsibilities on members of management, including the need to recruit and retain additional employees. Our financial performance and ability to compete will depend on our ability to manage growth effectively. To that end, we must:
• | manage our sales and marketing efforts, clinical trials, research and manufacturing activities effectively; |
• | hire more management, clinical development, administrative and sales and marketing personnel; and |
• | continue to develop our administrative systems and controls. |
Failure to accomplish any of these tasks could harm our business.
If we lose key personnel or are unable to attract more skilled personnel, we may be unable to pursue our product development and commercialization goals.
Our ability to operate successfully and manage growth depends upon hiring and retaining skilled managerial, scientific, sales, marketing, and financial personnel. The job market for qualified personnel is intensely competitive. We depend on the principal members of our management and scientific staff. Any officer or employee can terminate his or her relationship with us at any time and work for a competitor. We do not have employment insurance covering any of our personnel. The loss of key individuals could delay our research, development and commercialization efforts.
Risks Related to our Capital Needs and Financial Results
We may need additional capital to fund our operations or for strategic reasons. Such capital may not be available on acceptable terms or at all.
We are dependent on revenue from the sale of Korlym and our cash reserves to fund our commercial operations and development programs. If Korlym revenue declines, we may need to raise funds to support our plans. We may also choose to raise funds for strategic reasons. We cannot be certain funding will be available on acceptable terms or at all. In any event, equity financing would cause dilution and debt financing, if available, may involve restrictive covenants. If we obtain funds through collaborations with other companies, we may have to relinquish rights to Korlym or our product candidates. If adequate funds are not available, we may have to delay, reduce the scope of, or eliminate one or more of our development programs or even discontinue operations.
If we acquire products or product candidates, we will incur significant costs and may not realize the benefits we anticipate.
We may acquire a product or product candidate that complements our strategic plan. Such an acquisition may give rise to unforeseen difficulties and costs and may absorb significant management attention. We may not realize the anticipated benefits of any acquisition, which could dilute our stockholders’ ownership interest or cause us to incur significant expenses and debt.
18
If we are unable to obtain or maintain orphan designation for our product candidates our financial results may be negatively affected.
In the United States and the EU, orphan drug designation confers financial incentives such as opportunities for grant funding towards clinical trial costs, tax advantages, and reduction of fees or fee waivers. Although we have received orphan drug designation for relacorilant for the treatment of patients with Cushing’s syndrome and patients with pancreatic cancer in both the United States and EU, we may be unable to maintain these designations or to obtain designations for our other product candidates, which may negatively affect our financial results.
Risks Relating to Our Intellectual Property
To succeed, we must secure and maintain adequate patent protection for the composition and methods of use of our proprietary, selective cortisol modulators and for the use of Korlym to treat Cushing’s syndrome and other disorders.
Patents are uncertain, involve complex legal and factual questions and are frequently the subject of litigation. The patents issued or licensed to us may be challenged at any time. Similarly, competitors and others may take actions we believe infringe our intellectual property, causing us to take legal action to defend our rights. Litigating with respect to patents and other forms of intellectual property is lengthy, expensive and requires significant management attention. Outcomes are uncertain. If we do not protect our intellectual property, competitors may erode our competitive advantage. Please see “Part I, Item 3, Legal Proceedings.”
Our patent applications may not result in issued patents. Any patent issued to us may be challenged, invalidated, held unenforceable or circumvented. Our patent claims may not prevent third parties from producing competing products. The foreign countries in which we may someday operate may not protect our intellectual property to the extent the laws of the United States do. If we fail to obtain adequate patent protection in other countries, others may produce competing products in those countries based on our technology.
Third parties may allege that our patents infringe their rights. Defending against such allegations may result in costly litigation and may require us to obtain a license or bar us from commercializing our product candidates or Korlym for a new indication.
Our development and commercialization of Korlym or our selective cortisol modulators may give rise to claims that our patents or the patents we have licensed infringe the rights of others, which may require us to engage in costly, time-consuming and possibly unsuccessful litigation. If it is determined that one of our products or product candidates infringe others’ patent rights, we may have to obtain licenses to those rights or delay or suspend commercial activity while we attempt to design around the infringed patent. If our efforts fail, we may be unable to commercialize the infringing product or product candidate. We do not have liability insurance for patent infringement.
We do not believe that we infringe any patents or other proprietary rights. We are not obligated to pay royalties relating to the use of intellectual property except to the University of Chicago. To maintain these licenses, we must make milestone and royalty payments. If we do not comply with our payment and other obligations, we may lose the right to commercialize cortisol modulators, including mifepristone, for the treatment of TNBC and CRPC.
Our ability to compete could be diminished if we are unable to protect our trade secrets and proprietary information.
In addition to patents, we rely on a combination of confidentiality, nondisclosure and other contractual provisions, laws protecting trade secrets and security measures to protect our proprietary information. These measures may not be adequate, in which case competitors could exploit our proprietary information to our disadvantage. If employees, consultants or anyone else breaches their agreements with us regarding our proprietary information, we may not have adequate remedies for the breach.
The mifepristone patents we own or license cover the use of mifepristone, not its composition, which may make it harder to prevent patent infringement.
We own or have exclusively licensed issued U.S. patents covering the use of cortisol modulators, including mifepristone, to treat a variety of disorders. A method of use patent covers only a particular use of a compound, not its composition. Because our patents do not cover the composition of mifepristone, we cannot prevent others from commercializing mifepristone to treat disorders not covered by our method of use patents. The availability of mifepristone for these disorders may enable patients to obtain mifepristone from other companies for indications covered by our patents. Although such “off-label” use would violate our patents, effectively monitoring compliance and enforcing our rights may be difficult and costly. Mifepristone is sold in the United States by Danco Laboratories for the termination of pregnancy. We cannot be certain that patients with Cushing’s syndrome will not be able to obtain mifepristone from Danco or from another company, should it receive approval to market mifepristone for any indication.
19
Risks Related to Our Stock
The price of our common stock fluctuates widely and is likely to continue to do so. Opportunities for the sale of shares at any particular time may be limited.
We cannot assure investors that a liquid trading market for our common stock will exist at any particular time. As a result, holders of our common stock may not be able to sell shares quickly or at the current market price. During the 52-week period ended February 12, 2020, our average daily trading volume was approximately 814,063 shares and the intra-day sales prices per share of our common stock on The Nasdaq Stock Market ranged from $9.55 to $17.48. As of February 12, 2020, our officers, directors and principal stockholders beneficially owned approximately 16 percent of our common stock.
Our stock price can experience extreme price and volume fluctuations that are unrelated or disproportionate to our operating performance or prospects. Securities class action lawsuits are often instituted against companies following periods of stock market volatility. Such litigation is costly and diverts management’s attention from productive efforts.
Factors that may cause the price of our common stock to fluctuate rapidly and widely include:
• | changes in the expected or actual timing of our competitors’ potential development programs, including developments in ANDA litigation and proceedings before the PTAB and the announcement of ANDA filings seeking approval for generic versions of Korlym; |
• | actual or anticipated variations in our operating results or changes to any public guidance we have provided; |
• | actual or anticipated timing and results of our clinical trials; |
• | disputes or other developments relating to proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our technologies; |
• | short selling of our common stock, the publication of speculative opinions about our business or other market manipulation activities by third parties that are intended to lower our stock price or increase its volatility; |
• | changes in estimates or recommendations by securities analysts or the failure of our performance to meet the published expectations of those analysts or any public guidance we have provided; |
• | actual or anticipated regulatory approvals of our product candidates or of competing products; |
• | purchases or sales of our common stock by our officers, directors or stockholders; |
• | changes in laws or regulations applicable to our product candidates or our competitors’ products; |
• | technological innovations by us, our collaborators or our competitors; |
• | changes in the trading volume of our common stock; |
• | conditions in the pharmaceutical industries, including the market valuations of companies similar to Corcept; |
• | general market and economic conditions; |
• | additions or departures of key personnel; |
• | announcements by us or our competitors of significant acquisitions, strategic partnerships, joint ventures, collaborations or capital commitments; |
• | our cash and short-term investment position; and |
• | additional financing activities. |
Our stock price may decline if our financial performance does not meet the guidance that we provided to the public, estimates published by research analysts or other investor expectations.
The guidance we provide as to our expected 2020 revenue is only an estimate of what we believe is realizable at the time we give such guidance. Our actual results may vary materially. It is difficult to predict our revenue. For example, the rate of physician adoption of Korlym and the actions of government and private payers is uncertain. We may experience competition from generic versions of Korlym, which our public revenue guidance does not anticipate. We may not meet our financial guidance or other
20
investor expectations for other reasons, including those arising from the risks and uncertainties described in this report and in our other public filings and public statements. Research analysts publish estimates of our future revenue and earnings based on their own analysis. The revenue guidance we provide may be one factor they consider when determining their estimates.
Research analysts may not continue to provide or initiate coverage of our common stock or may issue negative reports.
The market for our common stock may be affected by the reports financial analysts publish about us. If any of the analysts covering us downgrades or discontinues coverage of our stock, the price of our common stock could decline rapidly and significantly. Paucity of research coverage may also adversely affect our stock price.
Sale of a substantial number of shares of our common stock may cause its price to decline.
Sales of a substantial number of shares of our stock in the public market could reduce its price. As additional shares of our stock become available for public resale, whether by the exercise of stock options by employees or directors or because of an equity financing by us, the supply of our stock will increase, which could cause its price to fall. Substantially all of the shares of our stock are eligible for sale, subject to applicable volume and other resale restrictions.
Our officers, directors and principal stockholders, acting as a group, could significantly influence corporate actions.
As of February 12, 2020, our officers and directors beneficially owned approximately 16 percent of our common stock. Acting together, these stockholders could significantly influence any matter requiring approval by our stockholders, including the election of directors and the approval of mergers or other business combinations. The interests of this group may not always coincide with our interests or the interests of other stockholders and may prevent or delay a change in control. This significant concentration of share ownership may adversely affect the trading price of our common stock because many investors perceive disadvantages to owning stock in companies with controlling stockholders.
Changes in laws and regulations may significantly increase our costs, which could harm our financial results.
New laws and regulations, as well as changes to existing laws and regulations, including statutes and regulations concerning the development, approval, and marketing of medications, the provisions of the PPACA requiring the reporting of aggregate spending related to health care professionals, the provisions of the Sarbanes-Oxley Act of 2002 and rules adopted by the SEC and by The Nasdaq Stock Market have and will likely continue to increase our cost of doing business and divert management’s attention from revenue-generating activities.
We may fail to comply with our public company obligations, including securities laws and regulations. Such compliance is costly and requires significant management attention.
The federal securities laws and regulations, including the corporate governance and other requirements of the Sarbanes-Oxley Act of 2002, impose complex and continually changing regulatory requirements on our operations and reporting. These developing requirements will continue to increase our compliance costs. Section 404 of the Sarbanes-Oxley Act of 2002 requires that we evaluate the effectiveness of, and provide a management report with respect to, our internal controls over financial reporting. It also requires that the independent registered public accounting firm auditing our consolidated financial statements must attest to and report on the effectiveness of our internal controls over financial reporting. If we are unable to complete the required assessment and report or if our independent registered public accounting firm is unable to issue an unqualified opinion as to the effectiveness of our internal control over financial reporting, investors could lose confidence in our financial reporting and our stock price would likely decline.
Anti-takeover provisions in our charter and bylaws and under Delaware law may make an acquisition of us or a change in our management more expensive or difficult, even if an acquisition or a management change would be beneficial to our stockholders.
Provisions in our charter and bylaws may delay or prevent an acquisition of us or a change in our management. Some of these provisions allow us to issue preferred stock without any vote or further action by the stockholders, require advance notification of stockholder proposals and nominations of candidates for election as directors and prohibit stockholders from acting by written consent. In addition, a supermajority vote of stockholders is required to amend our bylaws. Our bylaws provide that special meetings of the stockholders may be called only by our Chairman, President or the Board of Directors and that the authorized number of directors may be changed only by resolution of the Board of Directors. These provisions may prevent or delay a change in our Board of Directors or our management, which our Board of Directors appoints. In addition, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law. Section 203 may prohibit large stockholders, in particular those owning 15 percent or more of our outstanding voting stock, from merging or combining with us. These provisions in our charter and bylaws and under Delaware law could reduce the price that investors would be willing to pay for shares of our common stock.
21
ITEM 1B. UNRESOLVED STAFF COMMENTS
None.
ITEM 2. PROPERTIES
We lease 36,422 square feet of office space in Menlo Park, California for our corporate facilities. Our current lease expires in March 2022.
ITEM 3. LEGAL PROCEEDINGS
Teva ANDA Litigation.
On February 5, 2018, we received a Paragraph IV Notice Letter advising that Teva had submitted an Abbreviated New Drug Application (“ANDA”) to the FDA seeking authorization to manufacture, use or sell a generic version of Korlym in the United States prior to the expiration of certain of our patents related to Korlym - U.S. Patent No. 8,921,348 (the “’348 patent”) and U.S. Patent No. 9,829,495 (the “’495 patent”) - which are listed in the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations (referred to as the “Orange Book”). Teva’s February 5, 2018 Notice Letter alleges that the ’348 patent, with an expiration date in August 2028, and the ’495 patent, with an expiration date in August 2036, will not be infringed by Teva’s proposed product, are invalid and/or are unenforceable. On March 15, 2018, we filed a lawsuit in the U.S. District Court for the District of New Jersey against Teva for infringement of these patents. On October 12, 2018, Teva received tentative approval from the FDA for its ANDA. In accordance with the Hatch-Waxman Act, however, as a result of having filed a timely lawsuit against Teva, FDA final approval of Teva’s ANDA will be stayed until the earlier of (i) August 1, 2020 (i.e., 30 months from our February 1, 2018 receipt of Teva’s Paragraph IV Notice Letter) or (ii) a District Court decision finding that the identified patents are invalid, unenforceable or not infringed.
On July 6, 2018, we filed an Amended Complaint against Teva, asserting infringement of U.S. Patent No. 9,943,526 (the “’526 patent”). On February 8, 2019, we filed a second lawsuit against Teva, asserting infringement of U.S. Patent Nos. 10,166,242 (the “ʼ242 patent”), 10,166,243 (the “ʼ243 patent”) and 10,195,214 (the “ʼ214 patent”). On February 21, 2019, the District Court consolidated the two lawsuits. On December 13, 2019, we filed a third lawsuit against Teva, asserting infringement of U.S. Patent Nos. 10,500,216 (“the ʼ216 patent”).
No new 30-month stay results from the filing of the Amended Complaint or new lawsuits.
On May 7, 2019, Teva submitted to the PTAB a petition for post-grant review of the ’214 patent, which we opposed. On November 20, the PTAB granted Teva’s petition. A PTAB decision regarding the ‘214 patent is expected on or about November 20, 2020, subject to appeal to the United States Court of Appeals for the Federal Circuit.
We will vigorously enforce our intellectual property rights relating to Korlym, but cannot predict the outcome of these matters.
Sun ANDA Litigation
On June 10, 2019, we received a Paragraph IV Notice Letter advising that Sun had submitted an Abbreviated New Drug Application (“ANDA”) to the FDA seeking authorization to manufacture, use or sell a generic version of Korlym in the United States prior to the expiration of certain of our patents related to Korlym listed in the Orange Book (the “Korlym Patents”).
The Notice Letter alleges that the Korlym Patents will not be infringed by Sun Ltd.’s proposed product, are invalid and/or are unenforceable. On July 22, 2019, we filed a lawsuit in the U.S. District Court for the District of New Jersey against Sun Pharma Global FZE (“Sun FZE”), Sun Pharma Global Inc. (“Sun Pharma”), Sun Pharmaceutical Industries, Inc. (“Sun Inc.”), and Sun Ltd. (collectively, “Sun”) for infringement of the ’348, ’214, and ’495 patents. Sun has denied our allegations.
In accordance with the Hatch-Waxman Act, as a result of having filed a timely lawsuit against Sun, FDA approval of Sun Ltd.’s ANDA will be stayed until the earlier of (i) 30 months from our June 10, 2019 receipt of Sun Ltd.’s Paragraph IV Notice Letter or (ii) a District Court decision finding that the ’348, ’214, and ’495 patents are invalid, unenforceable or not infringed.
We will vigorously enforce our intellectual property rights relating to Korlym, but cannot predict the outcome of this matter.
Inter Partes Review at the PTAB
In August 2018, Neptune Generics, LLC (“Neptune”) submitted a petition for Inter Partes Review (“IPR”) at the PTAB of the ’348 patent. Neptune is backed by Burford Capital Ltd., a U.K.-based ligation finance company, and does not have regulatory approval to sell any drug in the United States. A PTAB decision finding all claims of the ‘348 patent to be valid was issued on
22
February 10, 2020. Neptune may petition the PTAB to reconsider its decision or may appeal the ruling to the Federal Circuit Court of Appeals. We would vigorously oppose either of these actions by Neptune.
Other matters
On March 14, 2019, a purported securities class action complaint was filed in the U.S. District Court for the Northern District of California by Nicholas Melucci (Melucci v. Corcept Therapeutics Incorporated, et al., Case No. 5:19-cv-01372-LHK). The complaint named us and certain of our executive officers as defendants asserting violations of Sections 10(b) and 20(a) of the Exchange Act and Rule 10b-5 promulgated thereunder and alleges that the defendants made false and materially misleading statements and failed to disclose adverse facts about our business, operations, and prospects. The complaint asserts a putative class period stemming from August 2, 2017 to February 5, 2019 and seeks unspecified monetary relief, interest and attorneys’ fees. On October 7, 2019, the Court appointed a lead plaintiff and lead counsel. The lead plaintiff’s consolidated complaint was filed on December 6, 2019. We moved to dismiss the class action complaint on January 27, 2020, but cannot predict the outcome of this matter.
On September 30, 2019, a purported shareholder derivative complaint was filed in the United States District Court for the District of Delaware by Lauren Williams, and captioned Lauren Williams v. G. Leonard Baker, et al., Civil Action No. 1:19-cv-01830. The complaint named our board of directors, including our Chief Executive Officer, as well as our Chief Financial Officer as defendants and us as nominal defendant. The complaint seeks to allege causes of action for breach of fiduciary duty, violation of Section 14(a) of the Exchange Act, insider selling and misappropriation of insider information, and waste of corporate assets. The complaint seeks an amount of damages to be proved at trial. On October 23, 2019, this action was stayed pending a resolution of the motion to dismiss filed in the securities class action. We will respond to this complaint vigorously but cannot predict the outcome of this matter.
On December 19, 2019, a second purported shareholder derivative complaint was filed in the United States District Court for the District of Delaware by Jeweltex Pension Plan, and captioned Jeweltex Pension Plan v. James N. Wilson, et al., Civil Action No. 1:19-cv-02308. The complaint named our board of directors, including our Chief Executive Officer, as well as our Chief Financial Officer as defendants and Corcept Therapeutics Incorporated as nominal defendant. The complaint seeks to allege causes of action for breach of fiduciary duty, violation of section 14(a) of the Exchange Act, waste of corporate assets, contribution and indemnification, aiding and abetting, and gross mismanagement. The complaint seeks an amount of damages to be proved at trial. We will respond to this complaint vigorously but cannot predict the outcome of this matter.
In addition to the matters described above, we are involved from time to time in other legal proceedings in the ordinary course of business. Although the outcome of any pending matters and the amount, if any, of our ultimate liability with respect to them cannot be predicted with certainty, we do not believe that the ultimate outcome of such matters will have a material adverse effect on our business, results of operations or financial position.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
23
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Our common stock is traded on The Nasdaq Capital Market under the symbol “CORT.”
Stockholders of Record and Dividends
As of February 12, 2020, we had 114,594,745 shares of common stock outstanding held by 27 stockholders of record. Because almost all of our common stock is held by brokers, nominees and other institutions on behalf of stockholders, we are unable to estimate the actual number of our stockholders. We have never declared or paid cash dividends. We do not anticipate paying cash dividends in the foreseeable future.
Sale of Unregistered Securities
None.
Repurchases of Securities
During the three months ended December 31, 2019, we paid approximately $1.9 million in employee withholding taxes due upon the vesting of, and related to, net settled equity awards. We withheld 0.2 million shares of common stock from employees to satisfy the related cost and statutory withholding requirements in connection with such net share settlement at an average price of $14.63 per share. These transactions may be deemed to be “issuer purchases” of shares.
Market Performance Graph
The graph and the accompanying text below is not “soliciting material,” is not deemed filed with the SEC and is not to be incorporated by reference in any filings by us under the Securities Act or the Exchange Act, whether made before or after the date hereof and irrespective of any general incorporation language in such filing.
We have elected to use the Nasdaq US Benchmark TR Index and Nasdaq Biotechnology Index (consisting of a group of 120 companies in the biotechnology sector, including us) for purposes of the performance comparison that appears below, which shows the cumulative stockholder return assuming the investment of $100 and the reinvestment of any dividends and is based on the returns of the component companies weighted according to their market capitalizations.
The graph shows the cumulative total stockholder return assuming the investment of $100 and the reinvestment of any dividends and is based on the returns of the component companies weighted according to their market capitalizations as of the end of the period for which returns are indicated. We have never paid dividends on our common stock.
The return shown in the graph below for our common stock is not necessarily indicative of future performance. We do not make or endorse any predictions as to future stockholder returns.
24
Five-Year Cumulative Total Returns of our Common Stock (CORT),
the Nasdaq US Benchmark TR Index (NQUSBT) and
the Nasdaq Biotechnology Index (NBI)
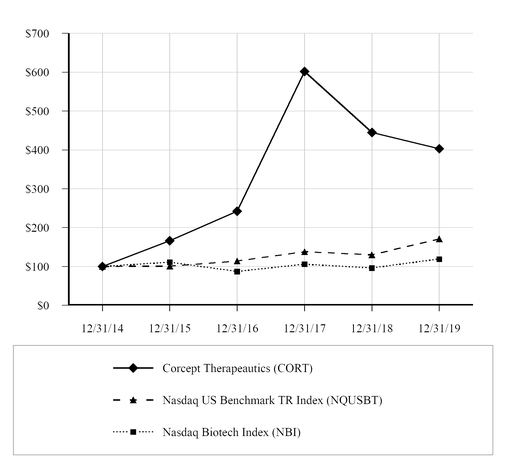
25
ITEM 6. SELECTED FINANCIAL DATA
SELECTED FINANCIAL DATA
(in thousands, except per share data)
The selected financial data set forth below are derived from our audited consolidated financial statements. The statement of operations data for the years ended December 31, 2019, 2018 and 2017 and the balance sheet data as of December 31, 2019 and 2018 are derived from our audited consolidated financial statements included in this Annual Report. The statement of operations data for the years ended December 31, 2016 and 2015 and the balance sheet data as of December 31, 2017, 2016 and 2015 have been derived from our audited financial statements, which are not included in this Annual Report. Our historical results are not necessarily indicative of our results for any future period. The selected financial data set forth below should be read in conjunction with our financial statements, the related notes and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included elsewhere in this Annual Report.
Year Ended December 31, | |||||||||||||||||||
2019 | 2018 | 2017 | 2016 | 2015 | |||||||||||||||
(In thousands, except per share data) | |||||||||||||||||||
Statement of Operations Data: | |||||||||||||||||||
Product revenue, net | $ | 306,486 | $ | 251,247 | $ | 159,201 | $ | 81,321 | $ | 50,286 | |||||||||
Operating expenses: | |||||||||||||||||||
Cost of sales | 5,504 | 5,215 | 3,554 | 2,058 | 1,361 | ||||||||||||||
Research and development | 89,017 | 75,247 | 40,376 | 23,844 | 15,419 | ||||||||||||||
Selling, general and administrative | 100,359 | 81,289 | 62,416 | 45,240 | 36,949 | ||||||||||||||
Total operating expenses | 194,880 | 161,751 | 106,346 | 71,142 | 53,729 | ||||||||||||||
Income (loss) from operations | 111,606 | 89,496 | 52,855 | 10,179 | (3,443 | ) | |||||||||||||
Interest and other income (expense), net | 5,070 | 2,657 | (49 | ) | (2,039 | ) | (2,965 | ) | |||||||||||
Income (loss) before income taxes | 116,676 | 92,153 | 52,806 | 8,140 | (6,408 | ) | |||||||||||||
Income tax expense (benefit) | 22,495 | 16,743 | (76,316 | ) | — | — | |||||||||||||
Net income (loss) | $ | 94,181 | $ | 75,410 | $ | 129,122 | $ | 8,140 | $ | (6,408 | ) | ||||||||
Net income (loss) per share: | |||||||||||||||||||
Basic | $ | 0.82 | $ | 0.65 | $ | 1.14 | $ | 0.07 | $ | (0.06 | ) | ||||||||
Diluted | $ | 0.77 | $ | 0.60 | $ | 1.04 | $ | 0.07 | $ | (0.06 | ) | ||||||||
Weighted average shares – basic | 114,349 | 115,343 | 113,527 | 110,566 | 106,883 | ||||||||||||||
Weighted average shares – diluted | 122,566 | 126,688 | 124,515 | 116,139 | 106,883 | ||||||||||||||
Includes certain non-cash expenses, of the following: | |||||||||||||||||||
Stock-based compensation | |||||||||||||||||||
Cost of sales | $ | 144 | $ | 259 | $ | — | $ | — | $ | — | |||||||||
Research and development | 9,541 | 7,012 | 3,743 | 1,312 | 839 | ||||||||||||||
Selling, general and administrative | 19,628 | 16,476 | 9,618 | 5,746 | 5,174 | ||||||||||||||
Total stock-based compensation | 29,313 | 23,747 | 13,361 | 7,058 | 6,013 | ||||||||||||||
Non-operating expense related to accretion of interest on long-term obligation | — | — | 456 | 1,929 | 2,848 | ||||||||||||||
Total non-cash expenses | $ | 29,313 | $ | 23,747 | $ | 13,817 | $ | 8,987 | $ | 8,861 | |||||||||
26
December 31, | |||||||||||||||||||
2019 | 2018 | 2017 | 2016 | 2015 | |||||||||||||||
(In thousands) | |||||||||||||||||||
Balance Sheet Data: | |||||||||||||||||||
Cash, cash equivalents and investments | $ | 315,314 | $ | 206,760 | $ | 104,025 | $ | 51,536 | $ | 40,435 | |||||||||
Working capital | 268,517 | 201,247 | 94,616 | 38,315 | 28,104 | ||||||||||||||
Total assets | 412,312 | 311,694 | 220,537 | 68,753 | 51,937 | ||||||||||||||
Debt obligation - current portion | — | — | — | 14,664 | 14,965 | ||||||||||||||
Debt obligation, net of current portion | — | — | — | — | 12,528 | ||||||||||||||
Total stockholders’ equity | 371,182 | 275,882 | 190,968 | 41,379 | 18,498 | ||||||||||||||
27
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following Management’s Discussion and Analysis of Financial Condition and Results of Operations (“MD&A”) is intended to help the reader understand our results of operations and financial condition and is provided as a supplement to, and should be read in conjunction with, our audited consolidated financial statements and the accompanying notes to financial statements, risk factors and other disclosures included in this Form 10-K. Our consolidated financial statements have been prepared in accordance with U.S. Generally Accepted Accounting Principles (“U.S. GAAP”).
We make statements in this section that are forward-looking statements within the meaning of the federal securities laws. For a complete discussion of such forward-looking statements and the potential risks and uncertainties that may affect their accuracy, see “Forward-Looking Statements” included in “Risk Factors” in this Form 10-K and the “Overview” and “Liquidity and Capital Resources” sections of this MD&A.
Overview
We are a commercial-stage company engaged in the discovery and development of drugs that treat severe metabolic, oncologic and psychiatric disorders by modulating the effects of the hormone cortisol. Since 2012, we have marketed Korlym® (mifepristone) for the treatment of patients who suffer from Cushing’s syndrome, a disease caused by excess cortisol activity.
We have discovered more than 500 proprietary, selective cortisol modulators in four structurally distinct series. Our lead compounds have entered the clinic as potential treatments for a variety of serious disorders - Cushing’s syndrome, solid tumors (including advanced, high-grade serous ovarian cancer, metastatic pancreatic cancer and castration-resistant prostate cancer), weight gain caused by antipsychotic medications, and non-alcoholic steatohepatitis (“NASH”).
Cushing’s Syndrome
Korlym. We sell Korlym in the United States, using experienced sales representatives to call on physicians caring for patients with endogenous Cushing’s syndrome (hypercortisolism). Because many people who suffer from Cushing’s syndrome are undiagnosed or inadequately treated, we have developed and continue to refine and expand programs to educate physicians and patients about screening for hypercortisolism and the role Korlym can play in treating the disorder. We also have a field-based force of medical science liaisons.
We use one specialty pharmacy and one specialty distributor to distribute Korlym and provide logistical support to physicians and patients. Our policy is that no patient with Cushing’s syndrome will be denied access to Korlym for financial reasons. To help us achieve that goal, we fund our own patient support programs and donate money to independent charitable foundations that help patients pay for all aspects of their Cushing’s syndrome care, whether or not that care includes taking Korlym.
Relacorilant. We are conducting a Phase 3 trial of our proprietary, selective cortisol modulator, relacorilant, as a treatment for hypercortisolism.
Relacorilant’s Phase 3 trial (“GRACE”), is expected to enroll 130 patients at sites in the United States, Canada, Europe and Israel. Each patient in GRACE will receive relacorilant for 22 weeks. Those who exhibit pre-specified improvements in hypertension or glucose metabolism will then enter a twelve-week, double-blind, “randomized withdrawal” phase, in which half of the patients will continue receiving relacorilant and the rest will receive placebo. GRACE’s primary endpoints are the rate and degree of relapse in patients receiving placebo compared to those continuing treatment with relacorilant.
We also plan to conduct a placebo-controlled, double-blind, Phase 3 trial of relacorilant to treat patients whose Cushing’s syndrome is caused by an adrenal tumor.
The FDA and the European Commission (“EC”) have designated relacorilant as an orphan drug for the treatment of Cushing’s syndrome. In the United States, orphan designation confers tax credits, reduced regulatory fees and, provided we obtain approval, seven years of exclusive marketing rights for relacorilant in the treatment of Cushing’s syndrome, with limited exceptions. Benefits of orphan drug designation by the EC are similar, and include reduced regulatory fees and, if we obtain approval, ten years of exclusive marketing rights in the European Union (“EU”) for the treatment of Cushing’s syndrome. Additional benefits in the EU include protocol assistance from the European Medicines Agency (“EMA”) and access to the EU's centralized marketing authorization procedure.
Oncology
Many types of solid tumors express GR and are potential targets for cortisol modulation therapy, among them pancreatic, ovarian, castration-resistant prostate and adrenocortical cancer.
28
Relacorilant in Patients with Solid Tumors. We are conducting a controlled Phase 2 trial of relacorilant in combination with Abraxane in patients with advanced, high-grade serous ovarian tumors. The trial is expected to enroll 180 patients at sites in the United States and Europe. Two thirds of the patients will receive relacorilant plus Abraxane. The rest will receive Abraxane alone. The primary endpoint is progression-free survival (“PFS”), as measured using the Response Evaluation Criteria in Solid Tumors (RECIST).
We plan to conduct a Phase 3 trial of relacorilant plus Abraxane to treat patients with metastatic pancreatic cancer. Relacorilant has been designated an orphan drug by both the FDA and the EC for the treatment of pancreatic cancer.
Cortisol Modulators in Patients with Castration-Resistant Prostate Cancer. We are conducting an open label, dose-finding trial of our proprietary, selective cortisol modulator exicorilant combined with Xtandi in patients with metastatic CRPC. Investigators at the University of Chicago are conducting a dose-finding trial of relacorilant combined with Xtandi in the same patient population. We are providing relacorilant. In addition to patents covering its composition of matter, we own United States patents covering the use of exicorilant to treat CRPC.
Metabolic Diseases
Antipsychotic-Induced Weight Gain and NASH. We are conducting a double-blind, placebo-controlled Phase 1b trial testing miricorilant's activity in attenuating antipsychotic-induced weight gain. The first part of this trial enrolled 66 healthy subjects, each of whom received ten mg per day of olanzapine and either placebo or miricorilant (600 mg). The duration of the trial was 14 days.
The trial’s second stage is testing a miricorilant dose of 900 mg. Planned enrollment is 30 healthy subjects.
We are conducting a Phase 2, double-blind, placebo-controlled trial of miricorilant in the reversal of antipsychotic-induced weight gain. We expect to enroll 100 patients with schizophrenia at 20 sites in the United States. Study participants will continue to receive their established antipsychotic medication and will have either miricorilant or placebo added to their regimen for 12 weeks. The trial’s primary endpoint is reduction in weight. We are also planning to conduct a double-blind placebo-controlled Phase 2 trial in patents with long-standing anti psychotic-induced weight gain.
Miricorilant is also potent in animal models of fatty liver and liver fibrosis. We plan to conduct a double-blind, placebo-controlled Phase 2 trial evaluating miricorilant as a treatment for NASH.
Continued Discovery and Development
We plan to continue identifying and developing proprietary, selective cortisol modulators.
Results of Operations
Net Product Revenue – Net product revenue is gross product revenue from sales to our customers less deductions for estimated government rebates and chargebacks.
Net product revenue was $306.5 million for the year ended December 31, 2019, compared to $251.2 million for the year ended December 31, 2018 and $159.2 million for the year ended December 31, 2017. The increases in net product revenue were primarily due to increased sales volume, as we shipped Korlym to more patients. Price increases represented approximately 41.6 percent, 14.3 percent and 16.6 percent of the increases in net revenue for the years ended December 31, 2019, 2018 and 2017, respectively. The increase in Korlym’s price for the year ended December 31, 2019 was due to a relative decrease in the number of patients covered by Medicaid (which reimburses Korlym at a lower rate), a statutorily-mandated increase in the price paid by other government programs, and a price increase that took effect on August 1, 2019.
Cost of sales – Cost of sales includes the cost of API, tableting, packaging, personnel, overhead, stability testing and distribution.
Cost of sales was $5.5 million for the year ended December 31, 2019, as compared to $5.2 million in 2018 and $3.6 million in 2017. For the year ended December 31, 2019, cost of sales was 1.8 percent of our net product revenue, as compared to 2.1 percent in 2018 and 2.2 percent in 2017. Cost of sales as a percentage of revenue declined due to an increase in the per-tablet price of Korlym. The dollar value of our cost of sales increased in both years due to greater sales unit volumes.
Research and development expenses – Research and development expenses include the cost of (1) recruiting and compensating development personnel, (2) clinical trials, (3) drug product and preclinical studies in support of clinical trials and regulatory submissions, (4) discovery research and (5) the development of drug formulations and manufacturing processes.
29
Research and development expenses increased to $89.0 million for the year ended December 31, 2019 from $75.2 million for the comparable period in 2018. The increase was primarily due to increased spending on the recruitment and compensation of development personnel and on the discovery and advancement of new selective cortisol modulators, partially offset by the completion of drug-drug interaction studies related to relacorilant.
Research and development expenses increased to $75.2 million for the year ended December 31, 2018 from $40.4 million in 2017, primarily due to the clinical advancement of relacorilant and pre-clinical and clinical development of miricorilant and exicorilant.
Year Ended December 31, | |||||||||||
2019 | 2018 | 2017 | |||||||||
(in thousands) | |||||||||||
Development programs: | |||||||||||
Oncology | $ | 21,098 | $ | 11,965 | $ | 7,465 | |||||
Endocrinology | 35,988 | 18,392 | 10,869 | ||||||||
Pre-clinical and clinical selective cortisol modulators | 11,120 | 29,380 | 13,605 | ||||||||
Unallocated activities, including pre-clinical, manufacturing and regulatory activities | 11,270 | 8,498 | 4,694 | ||||||||
Stock-based compensation | 9,541 | 7,012 | 3,743 | ||||||||
Total research and development expense | $ | 89,017 | $ | 75,247 | $ | 40,376 | |||||
It is difficult to predict the timing and cost of development activities, which are subject to many uncertainties and risks, including inconclusive or negative results, slow patient enrollment, adverse side effects and difficulties in the formulation or manufacture of study drugs and the lack of drug-candidate efficacy. In addition, clinical development is subject to intensive government oversight and regulations that may change unpredictably and without notice. We expect our research and development expense in 2020 to be higher than it was in 2019 as our clinical programs advance. Research and development spending in future years will depend on the outcome of our pre-clinical and clinical trials and our development plans.
Selling, general and administrative expenses - Selling, general and administrative expenses include (1) compensation of employees, consultants and contractors engaged in commercial and administrative activities, (2) the cost of vendors supporting commercial activities and (3) legal and accounting fees.
Selling, general and administrative expenses for the year ended December 31, 2019 increased to $100.4 million, from $81.3 million for the comparable period in 2018. The increases in selling, general and administrative expenses were primarily due to increased spending on the recruitment and compensation of additional employees, increased legal and marketing costs, and added distribution expenses arising from increased Korlym sales volumes.
Selling, general and administrative expenses for the year ended December 31, 2018 increased to $81.3 million, from $62.4 million for the comparable period in 2017. This increase was primarily due to increases in expenses for new and existing employees, volume-related pharmacy and other distribution costs and professional service fees.
We expect our selling, general and administrative expenses to be higher in 2020 than in 2019, due to increased commercial and administrative activities arising from increased sales volumes, intellectual property litigation and support for increased research and development activity. Selling, general and administrative activities in future years will depend on the cost and extent of our commercial activities and the scope of our research and development programs.
Interest and other income (expense), net - Interest and other income (expense), net for the year ended December 31, 2019 was $5.1 million, as compared to $2.7 million for the year ended December 31, 2018 and $(0.1) million for the year ended December 31, 2017. For the years ended December 31, 2019 and 2018, interest and other income primarily consisted of interest income from marketable securities, which increased in both years due to growth in our holdings of cash and marketable securities. For the year ended December 31, 2017, interest income from marketable securities was offset by interest expense arising from the that certain Purchase and Sale Agreement entered into with Biopharma in August 2012 (the “Financing Agreement”). We extinguished our obligations under the Financing Agreement in July 2017.
Income tax expense (benefit) - Income tax expense for the years ended December 31, 2019 and 2018 was $22.5 million and $16.7 million, respectively, and consisted primarily of our current statutory tax obligation offset by benefits from research and
30
development tax credits and exercises of stock options. The increase in income tax expense was primarily due to an increase in net income.
Income tax benefit for the year ended December 31, 2017 was $76.3 million, primarily due to recognition of the value of a portion of our accrued net operating losses and research and development tax credits. See Note 9, Income Taxes in our audited consolidated financial statements for additional information.
Liquidity and Capital Resources
Since 2015, we have relied on revenues from the sale of Korlym to fund our operations.
Based on our current plans, which include fully funding our Cushing’s syndrome commercial operations, conducting Phase 2 and Phase 3 trials of relacorilant in Cushing’s syndrome and solid tumors, the development of miricorilant to treat patients with antipsychotic-induced weight gain and NASH and of exicorilant to treat patients with CRPC, we expect to fund our operations without needing to raise additional funds, although we may choose to raise additional funds for other reasons. If we were to raise funds, equity financing would be dilutive to stockholders. Debt financing, if available, could involve restrictive covenants. Funds raised through collaborations with other companies may require us to relinquish certain rights in our product candidates.
At December 31, 2019, we had cash, cash equivalents and marketable securities of $315.3 million, consisting of cash and cash equivalents of $31.3 million and marketable securities of $284.0 million, compared to cash and cash equivalents of $41.6 million and marketable securities of $165.1 million at December 31, 2018.
The cash in our bank accounts and our marketable securities could be affected if the financial institutions holdings them were to fail or be subject to adverse conditions in the financial markets. We have never experienced a loss or lack of access to cash.
Net cash provided by operating activities for the year ended December 31, 2019, 2018 and 2017 was $136.1 million, $115.7 million and $60.9 million, respectively. These increases were primarily due to greater revenue.
Net cash used in investing activities for the years ended December 31, 2019, 2018 and 2017 was $117.8 million, $90.8 million and $73.5 million, respectively, primarily due to increased purchases of marketable securities with cash generated by our operating activities.
Net cash used in financing activities for the years ended December 31, 2019, 2018 and 2017 was $28.6 million, $14.3 million and $8.0 million respectively. For the same periods, stock option exercises provided $8.4 million, $9.3 million and $7.2 million, respectively. We repurchased an aggregate of $31.0 million and $23.7 million of our common stock during the years ended December 31, 2019 and 2018, respectively, pursuant to our program to repurchase up to $100 million of our common stock (the "Stock Repurchase Program"). During the year ended December 31, 2019, we also acquired 0.5 million shares at a cost of $6.1 million in satisfaction tax withholding requirements for the settlement of employee option exercises. We had no such transactions in 2018 and 2017. Because we extinguished the Financing Agreement in 2017, we made no payments under it in 2019 and 2018, compared to payments of $15.1 million during the year ended December 31, 2017.
We had an accumulated deficit of $23.6 million and $117.7 million in 2019 and 2018, respectively.
Contractual Obligations and Commitments
The following table presents our estimates of obligations under contractual agreements as of December 31, 2019.
Contractual Obligations | Total | Less than 1 year | 1-3 Years | 3-5 Years | More than 5 Years | ||||||||||||||
(in thousands) | |||||||||||||||||||
Manufacturing purchase commitments(1) | $ | 744 | $ | 744 | $ | — | $ | — | $ | — | |||||||||
Lease obligations(2) | $ | 4,662 | $ | 1,997 | $ | 2,665 | $ | — | $ | — | |||||||||
Research and development studies(3) | $ | 350 | $ | 350 | $ | — | $ | — | $ | — | |||||||||
Total other contractual obligations | $ | 5,756 | $ | 3,091 | $ | 2,665 | $ | — | $ | — | |||||||||
(1) As of December 31, 2019, we had commitments to purchase $0.6 million of API from PCAS.
(2) On October 23, 2019, we amended our office lease to add more space and extend its term. Effective October 1, 2019, the lease term was extended from March 31, 2020 through March 31, 2022 for the original office space and on April 1, 2020 the lease term will begin for the additional space through March 31, 2022. At December 31, 2019, the remaining minimum rental payments due under the lease were $4.7 million.
31
(3) In December 2013, we entered into an agreement with Quotient Sciences Limited (“Quotient”), a clinical research organization, to assist in the management and conduct of our Phase 1 studies of miricorilant and our other selective cortisol modulators. At December 31, 2019, the total non-cancelable commitment under the agreement was approximately $0.4 million.
We have other contractual payment obligations and purchase commitments, the timing of which are contingent on future events, including the initiation and completion of manufacturing projects. In March 2014, we entered into a long-term agreement with one contract manufacturer, PCAS to produce mifepristone, the API for Korlym. On July 25, 2018, we amended this agreement to add a second manufacturing site and extend its term to December 31, 2021, with two one-year automatic renewals, unless either party provides 12 months advance written notice of its intent not to renew. The amendment provides exclusivity between PCAS and Corcept. If PCAS is unable to meet our requirements, we may purchase mifepristone from another supplier.
We have agreements with two third-party manufacturers to produce and bottle Korlym tablets.
We enter into contracts in the normal course of business with CROs for preclinical studies and clinical trials. The contracts are cancellable, with varying provisions regarding termination. If a contract with a specific vendor were to be terminated, we would only be obligated for products and services we had received as of the effective date of the termination and any applicable cancellation fees.
Net Operating Loss Carryforwards
See Note 9, Income Taxes in our audited consolidated financial statements.
Off-Balance Sheet Arrangements
None.
Critical Accounting Policies and Estimates
Our consolidated financial statements have been prepared in accordance with U.S. GAAP, which requires us to make estimates and judgments that affect the amount of assets, liabilities and expenses we report. We base our estimates on historical experience and on other assumptions we believe to be reasonable. Actual results may differ from our estimates. Our significant accounting policies are described in Note 1, Basis of Presentation and Summary of Significant Accounting Policies, of the Notes to Consolidated Financial Statements included in Part IV of this Annual Report on Form 10-K. We believe the following accounting estimates and policies to be critical:
Net Product Revenue
To determine net product revenue, we deduct from sales the cost of our patient co-pay assistance program and our estimates of (i) government chargebacks and rebates, (ii) discounts provided to our SD for prompt payment and (iii) reserves for expected returns. We record these estimates at the time we recognize revenue and update them as new information becomes available. Our estimates take into account our understanding of the range of possible outcomes. If results differ from our estimates, we adjust our estimates, which changes our net product revenue and earnings. We report any changes in the period they become known to us, even if they concern transactions occurring in prior period.
Government Rebates
Korlym is eligible for purchase by, or qualifies for reimbursement from, Medicaid and other government programs that are eligible for rebates on the price they pay for Korlym. To determine the appropriate amount to reserve against these rebates, we identify Korlym sold to patients covered by government-funded programs, apply the applicable government discount to these sales and then estimate the portion of total rebates we expect will be claimed. We then (i) deduct this reserve from revenue in the period to which the rebates relate and (ii) include in accrued expenses on our consolidated balance sheet a current liability of equal amount.
Chargebacks
Although we sell Korlym to the SD at full price, some of the government entities to which the SD sells receive a discount. The SD recovers such discounts by reducing its payment to us (this reduction is called a “chargeback”). Chargebacks sometimes relate to Korlym sold to SD in prior periods. We deduct from our revenue in each period chargebacks claimed by the SD for Korlym we sold to the SD that period. We also create a reserve for chargebacks we estimate the SD will claim in future periods against Korlym it purchased in the current period but has not yet resold. We determine the amount of this reserve based on our experience with SD chargebacks and our understanding of the SD’s customer base and business practices. We deduct this reserve from revenue and include in accrued expenses on our consolidated balance sheet a current liability of equal amount.
32
Patient Assistance Program and Charitable Support
It is our policy that no patient be denied Korlym due to inability to pay. We provide financial assistance to eligible patients whose insurance policies have high deductibles or co-payments and deduct the amount of such assistance from gross revenue. We determine the assistance we provide each patient by applying our program guidelines to that patient’s financial position and their insurance policy’s co-payment and deductible requirements. We also donate cash to charities that help patients with financial need pay for the treatment of Cushing’s syndrome (which treatment may not include Korlym). We do not include in our revenue payments these charities make on behalf of patients receiving Korlym. We provide Korlym at no cost to patients without insurance who do not qualify for charitable support.
Sales Returns
For safety reasons, federal law prohibits patients from returning Korlym they have received. Korlym sold to our SD is subject to return. We deduct the amount of Korlym we estimate the SD will return from each period’s gross revenue. We base our estimates on quantitative and qualitative information including, but not limited to, historical return rates, the amount of Korlym held by the SD and projected demand. If we cannot reasonably estimate returns with respect to a particular sale, we defer recognition of revenue until we can make a reasonable estimate. To date, returns have not been material.
Leases
We adopted ASC Topic 842, effective January 1, 2019, using the modified retrospective method. The reported results for fiscal year 2019 reflect the application of ASC Topic 842, while the reported results for prior fiscal years are not adjusted and continue to be reported under ASC Topic 840. Refer to Recently Adopted Accounting Pronouncements in Part IV, Notes to Consolidated Financial Statements regarding the adoption impact of ASC Topic 842 in the year ended December 31, 2019.
We recognize right-of-use assets and lease liabilities at lease commencement. We measure lease liabilities based on the present value of lease payments over the lease term discounted by the rate equal to the rate we would pay on a loan with monthly payments and a term equal to the monthly payments and remaining term of our lease. We estimate our incremental borrowing rate based on bank quotes and an analysis of public companies with debt and credit carrying terms similar to our lease term. We do not include in the lease term options to extend or terminate the lease unless it is reasonably certain at commencement that we will exercise any such options. We account for the lease components separately from non-lease components for our operating leases.
Inventory and Cost of Sales
We value inventory at the lower of cost or net realizable value and determine the cost of inventory we sell using the specific identification method, which approximates a first-in, first-out basis. We assess our inventory levels at each reporting period and write down inventory that is either expected to be at risk of expiration prior to sale, or has a cost basis in excess of its expected net realizable value, or for which there are inventory quantities in excess of expected requirements. We destroy expired inventory and recognize the related costs as cost of sales in that period’s statement of comprehensive income.
Cost of sales includes the cost of manufacturing Korlym, including materials, third-party manufacturing costs and indirect personnel and other overhead costs, based on the number of Korlym tablets for which we recognize revenue, as well as costs of stability testing, logistics and distribution incurred during the applicable period.
Accruals of Research and Development Costs
We base our accruals for discovery research, preclinical studies and clinical trials on our estimates of work completed, milestones achieved, patient enrollment and past experience with similar activities. Our estimates include assessments of information from contract research organizations and the status of our own research, development and administrative activities.
Stock-based compensation
We account for stock-based compensation under the fair value method, based on the value of the award at the grant date. To date, our stock-based compensation has consisted entirely of option grants, which we value using the Black-Scholes model. We recognize stock-based compensation expense over the applicable vesting period, net of estimated forfeitures. If actual forfeitures differ from our estimates, we adjust stock-based compensation expense accordingly.
We recognize the expense of options granted to non-employees based on their fair value at the time of vesting.
33
Income Taxes
We account for income taxes in accordance with ASC 740, Income Taxes (“ASC 740”), which requires recognition of deferred tax assets and liabilities for the expected tax consequences of our future financial and operating activities. Under ASC 740, we determine deferred tax assets and liabilities based on the temporary difference between the financial statement and tax bases of assets and liabilities using the tax rates in effect for the year in which we expect such differences to reverse. If we determine that it is more likely than not that we will not generate sufficient taxable income to realize the value of some or all of our deferred tax assets (net of our deferred tax liabilities), we establish a valuation allowance offsetting the amount we do not expect to realize. We perform this analysis each reporting period and reduce or increase the size of our valuation allowance accordingly.
The deferred tax assets that we record each period depend primarily on our ability to generate future taxable income in the United States. Each period, we evaluate the need for a valuation allowance against our deferred tax assets and, if necessary, adjust the valuation allowance so that net deferred tax assets are recorded on our balance sheet only to the extent we conclude it is more likely than not that these deferred tax assets will be realized. If our outlook for future taxable income changes significantly, our assessment of the need for, and the amount of, a valuation allowance may also change.
We also account for uncertain tax positions in accordance with ASC 740, which requires us to adjust our consolidated financial statements to reflect only those tax positions that are more-likely-than-not to be sustained upon review by federal or state examiners. We recognize in the consolidated financial statements the largest expected tax benefit that has a greater than 50 percent likelihood of being sustained on examination by the taxing authorities. We report interest and penalties related to unrecognized tax benefits as income tax expenses.
Recently Issued Accounting Pronouncements
See Note 1, Basis of Presentation and Summary of Significant Accounting Policies in our audited consolidated financial statements.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Quantitative and Qualitative Disclosures About Market Risk
The primary objective of our investment activities is to preserve principal. As of December 31, 2019, the fair value of our cash and cash equivalents and marketable securities was $315.3 million. Our marketable securities consisted primarily of commercial paper, corporate notes, asset-backed securities, repurchase agreements, U.S. Treasury securities and a money market fund invested in short-term U.S. Treasury securities maintained at a major U.S. financial institution. To minimize our exposure to interest rate and other market risks, we have limited the maturities of our investments to less than three years, with the duration of our portfolio not to exceed two years. Due to the short-term nature and high liquidity of these instruments, an increase or decrease in market interest rates by 25 basis points would not have a material impact on the total value of our portfolio as of December 31, 2019.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
The consolidated financial statements required by this item are set forth beginning at page F-1 and are incorporated herein by reference.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
(a) Evaluation of Disclosure Controls and Procedures
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in the reports we file with the SEC is recorded, processed, summarized and filed within the time periods specified in the SEC’s rules and forms and that such information is accumulated and discussed with our management, including our Chief Executive Officer and Chief Financial Officer, so as to allow timely decisions regarding disclosure. Management recognizes that controls and procedures, no matter how well designed and operated, can only provide reasonable, not absolute, assurance the desired control objectives will be met. In reaching a reasonable level of assurance, management has weighed the cost of contemplated controls against their intended benefits. The design of any system of controls is based on management’s assumptions about the likelihood of future events. We cannot assure you that our controls will achieve their stated goals under all possible conditions. Changes in
34
future conditions may render our controls inadequate or may cause our degree of compliance with them to deteriorate. Because of the inherent limitations in a cost-effective control system, misstatements due to error or fraud may occur and not be detected.
As of December 31, 2019, our Chief Executive Officer and Chief Financial Officer evaluated our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) of the Exchange Act). Based on their evaluation, they concluded that they are effective.
There were no changes in our internal controls over financial reporting during the quarter ended December 31, 2019 that materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
(b) Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rule 13a-15(f). Our internal control system is designed to provide reasonable assurance regarding the preparation and fair presentation of externally-reported consolidated financial statements in accordance with U.S. GAAP. As discussed in Item 9A(a) above, internal control systems, no matter how well designed, have inherent limitations and can provide only reasonable assurance that their objectives have been met.
Our management, including our Chief Executive Officer and Chief Financial Officer, have evaluated the effectiveness of our internal control over financial reporting, based on criteria established in Internal Control – Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in 2013. Based on this evaluation, management concluded that, as of December 31, 2019, our internal control over financial reporting was effective.
Our independent registered public accounting firm has issued an attestation report on our internal control over financial reporting. It is set forth below.
(c) Report of Independent Registered Public Accounting Firm
To the Stockholders and Board of Directors of Corcept Therapeutics Incorporated
Opinion on Internal Control over Financial Reporting
We have audited Corcept Therapeutics Incorporated’s internal control over financial reporting as of December 31, 2019, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) (the COSO criteria). In our opinion, Corcept Therapeutics Incorporated (the Company) maintained, in all material respects, effective internal control over financial reporting as of December 31, 2019, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the consolidated balance sheets as of December 31, 2019 and 2018, the related consolidated statements comprehensive income, stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2019, and the related notes and our report dated February 24, 2020 expressed an unqualified opinion thereon.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects.
Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
35
Definition and Limitations of Internal Control Over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ Ernst & Young LLP
Redwood City, California
February 24, 2020
ITEM 9B. OTHER INFORMATION
None.
36
PART III
Certain information required by Part III is omitted from this Form 10-K because we expect to file with the U.S. Securities and Exchange Commission, not later than 120 days after the end of the fiscal year covered by this Annual Report on Form 10-K, a definitive proxy statement (“Proxy Statement”), pursuant to Regulation 14A in connection with the solicitation of proxies for our 2020 Annual Meeting of Stockholders, and certain information included therein is incorporated herein by reference.
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information required by this Item will be included in the Proxy Statement and is incorporated herein by reference.
ITEM 11. EXECUTIVE COMPENSATION
Compensation Discussion and Analysis
The information required by this Item will be included in the Proxy Statement and is incorporated herein by reference.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required by this Item will be included in the Proxy Statement and is incorporated herein by reference.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required by this Item will be included in the Proxy Statement and is incorporated herein by reference.
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
The information required by this Item will be included in the Proxy Statement and is incorporated herein by reference.
37
PART IV
ITEM 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULES
The following documents are filed as part of this Form 10-K
(1) Financial Statements:
Page | |
Audited Consolidated Financial Statements | |
(2) Financial Statement Schedules:
All schedules have been omitted because the information required to be set forth therein is not applicable or is shown in the financial statements or notes thereto.
(3) Exhibits:
Item 601 of Regulation S-K requires the exhibits listed below. Each management contract or compensatory plan or arrangement required to be filed as an exhibit to this Form 10-K has been identified.
(A) EXHIBITS
Exhibit Number | Description of Document |
3.1 | |
3.2 | |
4.1 | |
4.2 | |
4.3 | |
4.4 | |
4.5 | |
4.6 | |
38
Exhibit Number | Description of Document |
10.1 | |
10.2# | |
10.3† | |
10.4 | |
10.5† | |
10.6† | |
10.7† | |
10.8 | |
10.9† | |
10.10† | |
10.11† | |
10.12† | |
10.13# | |
10.14† | |
10.15 | |
10.16# | |
10.17# | |
10.18 | |
39
Exhibit Number | Description of Document |
10.19 | |
10.20 | |
10.21# | |
10.22 | |
10.23# | |
10.24 | |
10.25 | |
10.26# | |
10.27† | |
10.28† | |
10.29# | |
10.30# | |
10.31# | |
10.32† | |
10.33† | |
10.34 | |
10.35 | |
10.36 | |
10.37 | |
40
Exhibit Number | Description of Document |
10.38 | |
23.1 | |
24.1 | |
31.1 | |
31.2 | |
32.1 | |
32.2 | |
101.INS | XBRL Instance Document - the instance document does not appear in the Interactive Data File because its XBRL tags are embedded within the Inline XBRL document |
101.SCH | XBRL Schema Document |
101.CAL | XBRL Calculation Linkbase Document |
101.DEF | XBRL Definition Linkbase Document |
101.LAB | XBRL Labels Linkbase Document |
101.PRE | XBRL Presentation Linkbase Document |
104 | Cover Page Interactive Data File - the cover page interactive data file does not appear in the Interactive Data File because its XBRL tags are embedded within the Inline XBRL Document |
# | Confidential treatment granted |
† | Management contract or compensatory plan or arrangement |
ITEM 16. FORM 10-K SUMMARY
None.
41
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
CORCEPT THERAPEUTICS INCORPORATED | ||
By: | /s/ JOSEPH K. BELANOFF | |
Joseph K. Belanoff, M.D., | ||
Chief Executive Officer and President | ||
Date: | February 24, 2020 | |
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below hereby constitutes and appoints Joseph K. Belanoff and G. Charles Robb, and each of them acting individually, as his or her true and lawful attorneys-in-fact and agents, each with full power of substitution, for him or her in any and all capacities, to sign any and all amendments to this report on Form 10-K and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, with full power of each to act alone, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully for all intents and purposes as he or she might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents, or his or their substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Exchange Act, this Annual Report on Form 10-K has been signed by the following persons on behalf of the registrant and in the capacities and on the dates indicated:
Signature | Title | Date | ||
/s/ JOSEPH K. BELANOFF | Chief Executive Officer, President and Director | February 24, 2020 | ||
Joseph K. Belanoff, M.D. | (Principal Executive Officer) | |||
/s/ G. CHARLES ROBB | Chief Financial Officer and Secretary | February 24, 2020 | ||
G. Charles Robb | (Principal Financial Officer and Principal Accounting Officer) | |||
/s/ JAMES N. WILSON | Director and Chairman of the Board of Directors | February 24, 2020 | ||
James N. Wilson | ||||
/s/ G. LEONARD BAKER, JR. | Director | February 24, 2020 | ||
G. Leonard Baker, Jr. | ||||
/s/ DAVID L. MAHONEY | Director | February 24, 2020 | ||
David L. Mahoney | ||||
/s/ KIMBERLY PARK | Director | February 24, 2020 | ||
Kimberly Park | ||||
/s/ DANIEL N. SWISHER, JR | Director | February 24, 2020 | ||
Daniel N. Swisher, Jr. | ||||
42
CORCEPT THERAPEUTICS INCORPORATED
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
Page | ||
Audited Financial Statements | ||
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Stockholders and Board of Directors of Corcept Therapeutics Incorporated
Opinion on the Financial Statements
We have audited the accompanying consolidated balance sheets of Corcept Therapeutics Incorporated (the Company) as of December 31, 2019 and 2018, the related consolidated statements of comprehensive income, stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2019, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2019 and 2018, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2019, in conformity with U.S. generally accepted accounting principles.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company’s internal control over financial reporting as of December 31, 2019, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (2013 framework) and our report dated February 24, 2020 expressed an unqualified opinion thereon.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures include examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matter
The critical audit matter communicated below is a matter arising from the current period audit of the financial statements that was communicated or required to be communicated to the audit committee and that: (1) relates to accounts or disclosures that are material to the financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of the critical audit matter does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the account or disclosure to which it relates.
F-2
Inventory Excess and Obsolescence Reserve | |
Description of the Matter | As of December 31, 2019, the Company had $17.4 million of inventory which included $1.4 million of raw materials, $10.1 million of work in progress and $5.9 million of finished goods. As disclosed in Note 1, inventories are stated at the lower of cost or net realizable value. The Company assesses its inventory levels each reporting period and writes down inventory that is either expected to be at risk of expiration prior to sale, or has a cost basis in excess of its expected net realizable value, or for which there are inventory quantities in excess of expected requirements. Auditing management's estimates for excess and obsolete inventory involved subjective auditor judgment because the estimates rely on a number of factors that are affected by market and economic conditions outside the Company's control. In particular, the obsolete and excess inventory calculations are sensitive to significant assumptions, including the expected demand for the Company’s products, assumptions about the drug’s life cycle, the effect on demand of competitive products and the Company's purchase commitments. |
How We Addressed the Matter in Our Audit | We obtained an understanding, evaluated the design, and tested the operating effectiveness of internal controls over the Company's excess and obsolete inventory reserve process including management’s review of the significant assumptions described above and controls over the completeness and accuracy of the information used to develop the estimate. Our substantive audit procedures included, among others, evaluating methodologies used and data utilized in the analysis for inventory expected to be at risk for expiration or excess. We evaluated purchase commitments or alternative uses, compared forecasted demand to historical trends, compared actual inventory levels to forecasted demand requirements and evaluated the sensitivity of sales forecast assumptions on the amount of inventory reserves recorded. |
/s/ Ernst & Young LLP
We have served as the Company’s auditor since 2001.
Redwood City, California
February 24, 2020
F-3
CORCEPT THERAPEUTICS INCORPORATED
CONSOLIDATED BALANCE SHEETS
(In thousands, except per share data)
December 31, | |||||||
2019 | 2018 | ||||||
ASSETS | |||||||
Current assets: | |||||||
Cash and cash equivalents | $ | $ | |||||
Short-term marketable securities | |||||||
Trade receivables, net of allowances | |||||||
Inventory | |||||||
Prepaid expenses and other current assets | |||||||
Total current assets | |||||||
Strategic inventory | |||||||
Operating lease right-of-use asset | — | ||||||
Property and equipment, net of accumulated depreciation | |||||||
Long-term marketable securities | |||||||
Other assets | |||||||
Deferred tax assets, net | |||||||
Total assets | $ | $ | |||||
LIABILITIES AND STOCKHOLDERS' EQUITY | |||||||
Current liabilities: | |||||||
Accounts payable | $ | $ | |||||
Accrued clinical expenses | |||||||
Accrued and other liabilities | |||||||
Short-term operating lease liability | — | ||||||
Total current liabilities | |||||||
Long-term operating lease liability | — | ||||||
Long-term accrued income taxes | |||||||
Total liabilities | |||||||
Commitments and contingencies (Note 10) | |||||||
Stockholders’ equity: | |||||||
Preferred stock, par value $0.001 per share, 10,000 shares authorized and no shares outstanding at December 31, 2019 and December 31, 2018 | |||||||
Common stock, par value $0.001 per share, 280,000 shares authorized and 119,767 issued and 114,549 outstanding at December 31, 2019 and 116,838 shares issued and 115,031 outstanding at December 31, 2018 | |||||||
Treasury stock; at cost; 5,218 shares of common stock at December 31, 2019 and 1,807 shares of common stock at December 31, 2018 | ( | ) | ( | ) | |||
Additional paid-in capital | |||||||
Accumulated other comprehensive gain (loss) | ( | ) | |||||
Accumulated deficit | ( | ) | ( | ) | |||
Total stockholders’ equity | |||||||
Total liabilities and stockholders’ equity | $ | $ | |||||
The accompanying notes are an integral part of these consolidated financial statements.
F-4
CORCEPT THERAPEUTICS INCORPORATED
CONSOLIDATED STATEMENTS OF COMPREHENSIVE INCOME
(In thousands, except per share data)
Year Ended December 31, | |||||||||||
2019 | 2018 | 2017 | |||||||||
Product revenue, net | $ | $ | $ | ||||||||
Operating expenses: | |||||||||||
Cost of sales | |||||||||||
Research and development | |||||||||||
Selling, general and administrative | |||||||||||
Total operating expenses | |||||||||||
Income from operations | |||||||||||
Interest and other income (expense), net | ( | ) | |||||||||
Income before income taxes | 116,676 | ||||||||||
Income tax expense (benefit) | 22,495 | ( | ) | ||||||||
Net income | $ | $ | $ | ||||||||
Other comprehensive income (loss): | |||||||||||
Net unrealized gain (loss) on available-for-sale investments, net of tax impact of $(104), $22 and $0, respectively | ( | ) | |||||||||
Foreign currency translation gain, net of tax | — | — | |||||||||
Total comprehensive income | $ | $ | $ | ||||||||
Basic net income per share | $ | $ | $ | ||||||||
Diluted net income per share | $ | $ | $ | ||||||||
Weighted average shares outstanding used in computing net income per share | |||||||||||
Basic | |||||||||||
Diluted | |||||||||||
The accompanying notes are an integral part of these consolidated financial statements.
F-5
CORCEPT THERAPEUTICS INCORPORATED
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
Year Ended December 31, | |||||||||||
2019 | 2018 | 2017 | |||||||||
Cash flows from operating activities: | |||||||||||
Net income | $ | $ | $ | ||||||||
Adjustments to reconcile net income to net cash provided by operations: | |||||||||||
Stock-based compensation | |||||||||||
Accretion of interest (income) expense | ( | ) | ( | ) | |||||||
Depreciation and amortization of property and equipment | |||||||||||
Amortization of debt financing costs | |||||||||||
Deferred income taxes | ( | ) | |||||||||
Excess tax benefits from stock option activity | |||||||||||
Amortization of right-of-use asset | — | — | |||||||||
Changes in operating assets and liabilities: | |||||||||||
Trade receivables | ( | ) | ( | ) | ( | ) | |||||
Other receivable | ( | ) | |||||||||
Inventory | ( | ) | ( | ) | ( | ) | |||||
Prepaid expenses and other current assets | ( | ) | ( | ) | |||||||
Other assets | ( | ) | ( | ) | |||||||
Accounts payable | ( | ) | ( | ) | |||||||
Accrued clinical expenses | |||||||||||
Accrued and other liabilities | ( | ) | |||||||||
Long-term accrued income taxes | |||||||||||
Operating lease liability | ( | ) | |||||||||
Net cash provided by operating activities | |||||||||||
Cash flows from investing activities: | |||||||||||
Purchases of property and equipment | ( | ) | ( | ) | ( | ) | |||||
Proceeds from maturities of marketable securities | |||||||||||
Purchases of marketable securities | ( | ) | ( | ) | ( | ) | |||||
Net cash used in investing activities | ( | ) | ( | ) | ( | ) | |||||
Cash flows from financing activities: | |||||||||||
Proceeds from exercise of stock options, net of issuance costs | |||||||||||
Repurchase of common stock | ( | ) | ( | ) | |||||||
Payments related to debt obligation | ( | ) | |||||||||
Cash paid to satisfy statutory withholding requirement for the net settlement of cashless option exercise | ( | ) | — | — | |||||||
Net cash used in financing activities | ( | ) | ( | ) | ( | ) | |||||
Net increase (decrease) in cash and cash equivalents | ( | ) | ( | ) | |||||||
Cash and cash equivalents, at beginning of period | |||||||||||
Cash and cash equivalents, at end of period | $ | $ | $ | ||||||||
Supplemental disclosure: | |||||||||||
Income taxes paid | $ | $ | $ | ||||||||
Cost of shares repurchased for net settlement of cashless option exercise | $ | $ | $ | ||||||||
Recognition of right-of-use asset and lease liability | $ | $ | — | $ | — | ||||||
The accompanying notes are an integral part of these consolidated financial statements
F-6
CORCEPT THERAPEUTICS INCORPORATED
CONSOLIDATED STATEMENTS OF STOCKHOLDERS' EQUITY
(in thousands)
Common Stock | Additional Paid-in Capital | Treasury Stock | Accumulated Other Comprehensive Income (Loss) | Accumulated Deficit | Total Stockholders' Equity | |||||||||||||||||||||
Shares | Amount | |||||||||||||||||||||||||
Balance at December 31, 2016 | $ | $ | $ | $ | $ | ( | ) | $ | ||||||||||||||||||
Issuance of common stock upon exercise of options | — | — | — | |||||||||||||||||||||||
Stock-based compensation related to employee and director options | — | — | — | — | — | |||||||||||||||||||||
Stock-based compensation related to non-employee options | — | — | — | — | — | |||||||||||||||||||||
Other comprehensive loss, net of tax | — | — | — | — | ( | ) | — | ( | ) | |||||||||||||||||
Net income | — | — | — | — | — | |||||||||||||||||||||
Balance at December 31, 2017 | ( | ) | ( | ) | ||||||||||||||||||||||
Issuance of common stock upon exercise of options | — | — | — | |||||||||||||||||||||||
Stock-based compensation related to employee and director options | — | — | — | — | — | |||||||||||||||||||||
Other comprehensive income, net of tax | — | — | — | — | — | |||||||||||||||||||||
Purchase of treasury stock | ( | ) | — | — | ( | ) | — | — | ( | ) | ||||||||||||||||
Net income | — | — | — | — | — | |||||||||||||||||||||
Balance at December 31, 2018 | ( | ) | ( | ) | ( | ) | ||||||||||||||||||||
Issuance of common stock upon exercise of options | — | — | — | |||||||||||||||||||||||
Shares tendered to satisfy cost and statutory withholding requirements for net settlement of cashless option exercises | ( | ) | — | ( | ) | — | — | ( | ) | |||||||||||||||||
Stock-based compensation related to employee and director options | — | — | — | — | — | |||||||||||||||||||||
Stock-based compensation related to non-employee options | — | — | — | — | — | |||||||||||||||||||||
Other comprehensive income, net of tax | — | — | — | — | — | |||||||||||||||||||||
Purchases of treasury stock | ( | ) | — | — | ( | ) | — | — | ( | ) | ||||||||||||||||
Net income | — | — | — | — | — | |||||||||||||||||||||
Balance at December 31, 2019 | $ | $ | $ | ( | ) | $ | $ | ( | ) | $ | ||||||||||||||||
The accompanying notes are an integral part of these consolidated financial statements
F-7
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. Basis of Presentation and Summary of Significant Accounting Policies
Description of Business and Basis of Presentation
Corcept Therapeutics Incorporated is a commercial-stage pharmaceutical company engaged in the discovery and development of medications that treat severe metabolic, oncologic and psychiatric disorders by modulating the effect of the hormone cortisol. In 2012, the U.S. Food and Drug Administration (“FDA”) approved Korlym® (“mifepristone”) 300 mg tablets, as a once-daily oral medication for the treatment of hyperglycemia secondary to hypercortisolism in adult patients with endogenous Cushing’s syndrome who have type 2 diabetes mellitus or glucose intolerance and have failed surgery or are not candidates for surgery. We have discovered and patented four structurally distinct series of selective cortisol modulators, consisting of more than 500 compounds. We are developing compounds from these series as potential treatments for a broad range of serious disorders.
We were incorporated in the State of Delaware in May 1998. Our headquarters are located in Menlo Park, California.
Basis of Presentation
The consolidated financial statements have been prepared in accordance with U.S. generally accepted accounting principles (“U.S. GAAP”).
Principles of Consolidation
Our consolidated financial statements include the financial position and results of operations of Corcept Therapeutics UK Limited, our wholly owned subsidiary, which we incorporated in the United Kingdom in March 2017.
Use of Estimates
The preparation of consolidated financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the amounts reported in the consolidated financial statements and accompanying notes. Actual results could differ materially from those estimates.
We reevaluate our estimates and assumptions each quarter, including those related to revenue recognition, recognition and measurement of income tax assets and liabilities, inventory, allowances for doubtful accounts and other accrued liabilities, including our bonus accrual, clinical trial accruals and stock-based compensation.
Fair Value Measurements
We value financial instruments using assumptions we believe third-party market participants would use. When choosing which assumptions to make when determining the value of a financial instrument, we look first for quoted prices in active markets for identical instruments (“Level 1 inputs”). If no Level 1 inputs are available, we consider (i) quoted prices in non-active markets for identical instruments; (ii) active markets for similar instruments; (iii) inputs other than quoted prices for the instrument; and (iv) inputs that are not directly observable, but that can be corroborated by observable data (“Level 2 inputs”). In the absence of Level 2 inputs, we rely on unobservable inputs, such as our estimates of the assumptions market participants would use in pricing the instrument (“Level 3 inputs”).
Cash and Cash Equivalents and Marketable Securities
We consider highly liquid investments that will mature in three months or less from the time we purchase them to be cash equivalents. Cash equivalents are valued using Level 1 inputs, which approximate our cost.
We invest the majority of our funds in marketable securities, primarily corporate notes, U.S. Treasury securities, asset-backed securities, commercial paper and repurchase agreements. We classify our marketable securities as available-for-sale securities and report them at fair value as “cash equivalents” or “marketable securities” on our consolidated balance sheet, with related unrealized gains and losses included in stockholders' equity. Realized gains and losses and permanent declines in value are included in “interest and other income (expense)” on our consolidated statement of comprehensive income.
Credit and Concentration Risks
Our cash, cash equivalents and marketable securities are held in one financial institution. We are subject to credit risk from our cash equivalents and marketable securities. We limit our investments to U.S. Treasury obligations and high-grade corporate
F-8
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, Continued
debt, asset-backed securities and repurchase agreements with less than a 36-month maturity at the time of purchase. These investments are diversified and do not expose us to concentrations of credit risk. We have never experienced a loss in, or lack of access to, our operating or investment accounts.
We have a single-source manufacturer of mifepristone, the active pharmaceutical ingredient (API), in Korlym - Produits Chimiques Auxiliaires et de Synthèse SA (PCAS). If PCAS is unable or unwilling to manufacture API in the amounts and time frames required, we may not be able to manufacture Korlym in a timely manner. In order to mitigate this risk, we have purchased and hold in inventory a reserve quantity of mifepristone API.
We have a concentration of risk in regard to the distribution of our product. A single specialty pharmacy, Optime Care, Inc. (“Optime”), dispenses Korlym to patients for us. Optime is an independent third party. Its unwillingness or inability to dispense Korlym to patients in a timely manner would harm our business.
We sell the Korlym that Optime dispenses directly to patients, with title to the medicine passing directly from us to the patient upon the patient’s receipt of the drug. Our receivables risk is spread among various third-party payers - pharmacy benefit managers, insurance companies, government programs and private charities. We extend credit to third-party payers based on their creditworthiness. We monitor our exposure and record an allowance against uncollectible trade receivables as necessary. To date, we have not incurred any credit losses.
Inventory and Cost of Sales
Regulatory approval of product candidates is uncertain. Because product manufactured prior to regulatory approval may not be sold unless regulatory approval is obtained, we record the cost of manufacturing our product candidates as research and development expenses at the time such costs are incurred. We capitalize to inventory manufacturing costs related to Korlym.
We value inventory at the lower of cost or net realizable value and determine the cost of inventory we sell using the specific identification method, which approximates a first-in, first-out basis. We assess our inventory levels at each reporting period and write down inventory that is either expected to be at risk of expiration prior to sale, or has a cost basis in excess of its expected net realizable value, or for which there are inventory quantities in excess of expected requirements. We destroy expired inventory and recognize the related costs as cost of sales in that period’s statement of comprehensive income.
Cost of sales also includes the cost of manufacturing Korlym, including materials, third-party manufacturing costs and indirect personnel and other overhead costs, based on the number of Korlym tablets for which we recognize revenue, as well as costs of stability testing, logistics and distribution.
We classify inventory we do not expect to sell or use in clinical studies within 12 months of the balance sheet date as strategic inventory, a non-current asset.
Net Product Revenue
We sell Korlym directly to patients through a single specialty pharmacy. We also sell Korlym to a specialty distributor (“SD”), for which we recognize revenue at the time the SD receives Korlym. SD sales were less than one percent of our net revenue in the years ended December 31, 2019 and December 31, 2018.
To determine our revenue from the sale of Korlym, we (i) identify our contract with each customer; (ii) identify the obligations of Corcept and the customer under the contract; (iii) determine the contracted transaction price; (iv) allocate the transaction price to the contract’s performance obligations, which in our case consists of delivering Korlym to the customer; and (v) recognize revenue once Korlym has been delivered, provided we deem it probable that we will collect the payment due to us.
Confirmation of coverage by private or government insurance or by a third-party charity is a prerequisite for selling Korlym to a patient.
To determine net product revenue, we deduct from sales the cost of our patient co-pay assistance program and our estimates of (a) government chargebacks and rebates, (b) discounts provided to our SD for prompt payment and (c) reserves for expected Korlym returns. We record these estimates at the time we recognize revenue and update them as new information becomes available. Our estimates take into account our understanding of the range of possible outcomes. If results differ from our estimates, we adjust our estimates, causing a change to our net product revenue and earnings. We report any changes in the period they become known, even if they concern transactions occurring in prior periods.
Government Rebates: Korlym is eligible for purchase by, or qualifies for reimbursement from, Medicaid and other government programs that are eligible for rebates on the price they pay for Korlym. To determine the appropriate amount to reserve
F-9
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, Continued
against these rebates, we identify Korlym sold to patients covered by government-funded programs, apply the applicable government discount to these sales, then estimate the portion of total rebates we expect will be claimed.
Chargebacks. Although we sell Korlym to the SD at full price, some of the government entities to which the SD sells receive a discount. As it makes such sales, SD recovers the full amount of any related discounts by reducing its payment to us (this reduction is called a “chargeback”). Chargebacks sometimes relate to Korlym purchased by the SD in prior periods. We deduct from our revenue in each period chargebacks claimed by the SD for Korlym it purchased in that period. We also create each period a reserve for chargebacks we estimate the SD will claim in future periods against Korlym it has not yet resold. We determine the amount of this reserve based on our experience with SD chargebacks and our understanding of the SD’s customer base and business practices. We then deduct this reserve from revenue and include in accrued expenses on our consolidated balance sheet a current liability of equal amount.
Patient Assistance Program and Charitable Support: It is our policy that no patient be denied Korlym due to inability to pay. We provide financial assistance to eligible patients whose insurance policies have high deductibles or co-payments and deduct the amount of such assistance from gross revenue. We determine the assistance we provide each patient by applying our program guidelines to that patient’s financial position and their insurance policy’s co-payment and deductible requirements for the purchase of Korlym. We donate cash to charities that help patients with financial need pay for the treatment of Cushing’s syndrome. We do not include payments from these charities in revenue. We provide Korlym at no cost to uninsured patients who do not qualify for charitable support.
Sales Returns: Federal law prohibits the return of Korlym sold to patients. Sales to our SD are subject to return. We deduct the amount of Korlym we estimate the SD will return from each period’s gross revenue. We base our estimates on quantitative and qualitative information including, but not limited to, historical return rates, the amount of Korlym held by the SD and projected demand. If we cannot reasonably estimate returns with respect to a particular sale, we defer recognition of revenue until we can make a reasonable estimate. To date, returns have not been significant.
The following table summarizes activity in each of the product revenue allowance and reserve categories for the year ended December 31, 2019:
Chargebacks | Government Rebates | Total | |||||||||
(in thousands) | |||||||||||
Balance at December 31, 2016: | $ | $ | $ | ||||||||
Provision recorded during the period | |||||||||||
Credit or payments made during the period | ( | ) | ( | ) | ( | ) | |||||
Balance at December 31, 2017: | |||||||||||
Provision related to current period sales | |||||||||||
Provision related to prior period sales | |||||||||||
Credit or payments made during the period | ( | ) | ( | ) | ( | ) | |||||
Balance at December 31, 2018: | |||||||||||
Provision related to current period sales | |||||||||||
Provision related to prior period sales | ( | ) | ( | ) | |||||||
Credit or payments made during the period | ( | ) | ( | ) | ( | ) | |||||
Balance at December 31, 2019: | $ | $ | $ | ||||||||
Leases
We adopted ASC Topic 842, effective January 1, 2019, using the modified retrospective method. The reported results for fiscal year 2019 reflect the application of ASC Topic 842, while the reported results for prior fiscal years are not adjusted and continue to be reported under ASC Topic 840. Refer to Recently Adopted Accounting Pronouncements regarding the adoption impact of ASC Topic 842 in the year ended December 31, 2019.
We determine whether an arrangement contains a lease at inception. A contract is or contains a lease if the contract conveys the right to control the use of an identified asset for a period of time in exchange for consideration. To determine whether a contract
F-10
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, Continued
is or contains a lease, we consider all relevant facts and circumstances to assess whether the customer has the right to both (i) obtain substantially all of the economic benefits from use of the identified asset and (ii) direct the use of the identified asset.
We recognize right-of-use assets and lease liabilities at lease commencement. We measure lease liabilities based on the present value of lease payments over the lease term discounted using the rate equal to the rate we would pay on a loan with monthly payments and a term equal to the monthly payments and remaining term of our lease. We estimate our incremental borrowing rate based on non-tender bank quotes and an analysis of public companies with debt and credit carrying terms similar to our lease term. We do not include in the lease term options to extend or terminate the lease unless it is reasonably certain at commencement that we will exercise any such options. We account for the lease components separately from non-lease components for our operating leases.
We measure right-of-use assets based on the corresponding lease liabilities adjusted for (i) prepayments made to the lessor at or before the commencement date, (ii) initial direct costs we incur, and (iii) tenant incentives under the lease. We evaluate the recoverability of our right-of-use assets for possible impairment in accordance with our long-lived assets policy. We do not recognize right-of-use assets or lease liabilities for leases with a term of twelve months or less; rather, we recognize the associated lease payments in the consolidated statements of comprehensive income on a straight-line basis over the lease term.
Operating leases are reflected on our consolidated balance sheets as operating lease right-of-use assets, short-term operating lease liabilities and long-term operating lease liabilities.
We begin recognizing operating lease expense when the lessor makes the underlying asset available to us. We recognize operating lease expense under our operating leases on a straight-line basis. Variable lease payments are expensed as incurred.
Research and Development
Research and development expenses include the direct cost of discovering and screening new compounds, pre-clinical studies, clinical trials, manufacturing development, submissions to regulatory agencies and related overhead costs. We expense nonrefundable payments and the cost of technologies and materials used in research and development as we incur them.
Segment Reporting
We determine our operating segments based on the way we organize our business, make decisions and assess performance. We have only one operating segment, which is the discovery, development and commercialization of pharmaceutical products.
Stock-Based Compensation
We account for stock-based compensation under the fair value method, based on the value of the award at the grant date. To date, our stock-based compensation has consisted entirely of option grants, which we value using the Black-Scholes model. We recognize stock-based compensation expense over the applicable vesting period, net of estimated forfeitures. If actual forfeitures differ from our estimates, we adjust stock-based compensation expense accordingly.
We recognize the expense of options granted to non-employees based on their fair value at the time of vesting.
Income Taxes
We account for income taxes in accordance with ASC 740, Income Taxes (“ASC 740”), which requires recognition of deferred tax assets and liabilities for the expected tax consequences of our future financial and operating activities. Under ASC 740, we determine deferred tax assets and liabilities based on the temporary difference between the financial statement and tax bases of assets and liabilities using the tax rates in effect for the year in which we expect such differences to reverse. If we determine that it is more likely than not that we will not generate sufficient taxable income to realize the value of some or all of our deferred tax assets (net of our deferred tax liabilities), we establish a valuation allowance offsetting the amount we do not expect to realize. We perform this analysis each reporting period and reduce our measurement of deferred taxes, if the likelihood we will realize them becomes uncertain.
The deferred tax assets we record each period depend primarily on our ability to generate future taxable income in the United States. Each period, we evaluate the need for a valuation allowance against our deferred tax assets and, if necessary, adjust the valuation allowance so that net deferred tax assets are recorded only to the extent we conclude it is more likely than not that
F-11
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, Continued
these deferred tax assets will be realized. If our outlook for future taxable income changes significantly, our assessment of the need for, and the amount of, a valuation allowance may also change.
We are also required to evaluate and quantify other sources of taxable income, such as the possible reversal of future deferred tax liabilities, should any arise, and the implementation of tax planning strategies. Evaluating and quantifying these amounts is difficult and involves significant judgment, based on all of the available evidence and assumptions about our future activities.
We account for uncertain tax positions in accordance with ASC 740, which requires us to adjust our consolidated financial statements to reflect only those tax positions that are more-likely-than-not to be sustained upon review by federal or state examiners. We recognize in the consolidated financial statements the largest expected tax benefit that has a greater than 50 percent likelihood of being sustained on examination by the taxing authorities. We report interest and penalties related to unrecognized tax benefits as income tax expenses.
Recently Adopted Accounting Pronouncements
In February 2016, the FASB issued ASU No. 2016-02, “Leases”, which requires lease transactions with terms longer than 12 months to be recognized on the balance sheet as a liability (“lease liabilities”), offset by an asset of equal amount (“right-of-use assets”). ASU No. 2016-02 supersedes the lease accounting requirements of ASC Topic 840, “Leases” and creates Topic 842, “Leases.” We adopted this standard on January 1, 2019, using the modified retrospective approach, which did not cause adjustments to prior comparative periods. We have reviewed all of our contracts that may contain leases and have determined that the only impact is to the accounting for our leased office space. We have applied the practical expedients in Topic 842 that allow us not to reassess lease classification for expired or existing lease contracts. On the date of adoption, we increased our “operating right-of-use assets" and “operating lease liability” by approximately $1.9 million, an amount equal to the present value of our expected payments over the remaining term of the lease. There was no change to our retained earnings. See Note 5 for more information regarding our leased office space and additional operating right-of-use assets capitalized after the date of adoption.
In February 2018, the FASB issued ASU No. 2018-02, “Income Statement - Reporting Comprehensive Income (Topic 220): Reclassification of Certain Tax Effects from Accumulated Other Comprehensive Income.” This standard allows companies to reclassify to retained earnings tax effects related to items that have been stranded in “accumulated other comprehensive income” as a result of the Tax Cuts and Jobs Act (the “Act”). A company that elects to reclassify these amounts must reclassify stranded tax effects related to the Act’s change in US federal tax rate for all items accounted for in “other comprehensive income.” These entities can also elect to reclassify other stranded effects that relate to the Act but do not directly relate to the change in the federal rate. This standard is effective for fiscal years, and interim periods within those years, beginning after December 15, 2018. We adopted this standard on January 1, 2019. It had no impact on our consolidated financial statements.
In June 2018, the FASB issued ASU No. 2018-07, “Stock Compensation (Topic 718): Improvements to Nonemployee Share-Based Payment Accounting,” which expands the scope of ASC 718 to include all share-based payment arrangements related to the acquisition of goods and services from nonemployees. This standard is effective for fiscal years and interim periods within those years beginning after December 15, 2018. We adopted this standard on January 1, 2019. It had no impact on our consolidated financial statements.
Recently Issued Accounting Pronouncements Not Yet Adopted
In June 2016, the FASB issued ASU No. 2016-13, “Financial Instruments-Credit Losses (Topic 326), Measurement of Credit Losses on Financial Instruments,” which changes the methodology for measuring credit losses on financial instruments and when such losses are recorded. This standard is effective for fiscal years, and interim periods within those years, beginning after December 15, 2019. We will adopt this standard on January 1, 2020 using the modified retrospective approach with the cumulative effect of the adoption recorded as an adjustment to retained earnings. The effect on our consolidated financial statements and related disclosures is not expected to be material.
In August 2018, the FASB issued ASU No. 2018-13, “Fair Value Measurements (Topic 820),” which eliminates or modifies certain disclosure requirements for fair value measurements and requires disclosure of new information. This standard is effective for fiscal years, and interim periods within those years, beginning after December 15, 2019. We will adopt this standard on January 1, 2020 using the modified retrospective approach with the cumulative effect of the adoption recorded as an adjustment to retained earnings. The effect on our consolidated financial statements and related disclosures is not expected to be material.
In August 2018, the FASB issued ASU No. 2018-15, “Intangibles-Goodwill and Other-Internal-Use Software (Subtopic 350-40): Customer’s Accounting for Implementation Costs Incurred in a Cloud Computing Arrangement That Is a Service Contract,” which requires a customer that is a party to a cloud computing service contract to follow the internal-use software guidance in ASC 350-40 to determine which implementation costs to recognize as deferred assets. This standard is effective for
F-12
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, Continued
fiscal years, and interim periods within those years, beginning after December 15, 2019. We will adopt this standard on January 1, 2020 using the modified retrospective approach with the cumulative effect of the adoption recorded as an adjustment to retained earnings. The effect on our consolidated financial statements and related disclosures is not expected to be material.
In December 2019, the FASB issued ASU 2019-12 (ASC Topic 740), “Simplifying the Accounting for Income Taxes”. This standard simplifies accounting for income taxes by removing certain exceptions to the general principles and amending existing guidance to improve consistent application. This standard will be effective for fiscal years, and interim periods within those years, beginning after December 15, 2021. Early adoption is permitted. We are in the process of assessing the impact of this standard on our consolidated financial statements.
2. Significant Agreements
Commercial Agreements
In August 2017, we entered into a distribution services agreement with an independent third party, Optime, to provide exclusive specialty pharmacy and patient services programs for Korlym beginning August 10, 2017. Under the terms of this agreement, Optime acts as the exclusive specialty pharmacy distributor of Korlym in the United States, subject to certain exceptions. Optime provides services related to pharmacy operations; patient intake, access and reimbursement; patient support; claims management and accounts receivable; and data and reporting. We provide Korlym to Optime, which it dispenses to patients. Optime does not purchase Korlym from us and it does not take title to the product. Title passes directly from us to the patient at the time the patient receives the medicine.
The initial term of our agreement with Optime is five years , unless terminated earlier by us upon 90 days’ notice. The agreement contains additional customary termination provisions, representations, warranties and covenants. Subject to certain limitations, we have agreed to indemnify Optime for certain third-party claims related to the product, and we have each agreed to indemnify the other for certain breaches of representations, warranties, covenants and other specified matters.
Manufacturing Agreements Related to Korlym
We purchase all of our API for Korlym from PCAS. On July 25, 2018, we amended our agreement with PCAS to add a second manufacturing site and extend its term to December 31, 2021, with two one-year automatic renewals, unless either party provides 12 months advance written notice of its intent not to renew. The amendment provides exclusivity between PCAS and Corcept. In the event PCAS cannot meet our requirements, we may purchase API from another supplier. As of December 31, 2019, we had non-cancelable commitments to purchase $0.6 million worth of API from PCAS over the next 12 months.
We have agreements with two third-party manufacturers to produce and bottle Korlym tablets.
Research and Development Agreements
Our clinical trials are conducted through the use of clinical research organizations (“CROs”). Our Phase 3 GRACE trial of relacorilant for the treatment of patients with Cushing’s syndrome is being conducted under an agreement with ICON plc (“ICON”). IQVIA (formerly, "Novella Clinical LLC") is helping us conduct our Phase 2 trial of relacorilant to treat patients with metastatic ovarian cancer and our Phase 1/2 trial of exicorilant to treat patients with CRPC. Medpace, Inc. ("Medpace") is helping us conduct our Phase 2 trial testing miricorilant's activity in reversing recent antipsychotic-induced weight gain. Our agreements with ICON and IQVIA may be terminated by us on 60 days’ written notice or sooner if the parties mutually agree. Our agreement with Medpace may be terminated by us without cause at any time.
In July 2019, we entered into clinical study agreements with Quotient Sciences for clinical research on CORT113176, miricorilant and exicorilant, with initial terms of less than one year, with no extensions. We may terminate any of these agreements early should the study data justify or require termination. As of December 31, 2019, we had non-cancelable purchase commitments of approximately $0.4 million from Quotient over the next 12 months.
Lease Agreement
3. Available for Sale Securities and Fair Value Measurements
The available-for-sale securities in our Consolidated Balance Sheets are as follows:
F-13
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, Continued
Year Ended December 31, | |||||||
2019 | 2018 | ||||||
(in thousands) | |||||||
Cash equivalents | $ | $ | |||||
Short-term marketable securities | |||||||
Long-term marketable securities | |||||||
Total marketable securities | $ | $ | |||||
The following table presents our available-for-sale securities grouped by asset type:
Fair Value Hierarchy Level | December 31, 2019 | December 31, 2018 | |||||||||||||||||||||||||||||||
Amortized Cost | Gross Unrealized Gains | Gross Unrealized Losses | Estimated Fair Value | Amortized Cost | Gross Unrealized Gains | Gross Unrealized Losses | Estimated Fair Value | ||||||||||||||||||||||||||
(in thousands) | |||||||||||||||||||||||||||||||||
Corporate bonds | Level 2 | $ | $ | $ | ( | ) | $ | $ | $ | $ | ( | ) | $ | ||||||||||||||||||||
Commercial paper | Level 2 | ||||||||||||||||||||||||||||||||
Asset-backed securities | Level 2 | ( | ) | ( | ) | ||||||||||||||||||||||||||||
Repurchase agreements | Level 2 | ||||||||||||||||||||||||||||||||
U.S. treasury securities | Level 1 | ( | ) | ||||||||||||||||||||||||||||||
Money market funds | Level 1 | ||||||||||||||||||||||||||||||||
Total Marketable securities | $ | $ | $ | ( | ) | $ | $ | $ | $ | ( | ) | $ | |||||||||||||||||||||
We estimate the fair value of marketable securities classified as Level 1 using quoted market prices for these or similar investments obtained from a commercial pricing service. We estimate the fair value of marketable securities classified as Level 2 using inputs that may include benchmark yields, reported trades, broker/dealer quotes and issuer spreads.
We do not intend to sell the investments that are currently in an unrealized loss position, and it is highly unlikely that we will be required to sell the investments before recovery of their amortized cost basis, which may be maturity.
As of December 31, 2019, all our marketable securities had original maturities of less than two years . The weighted-average maturity of our holdings was six months . As of December 31, 2019, our long-term marketable securities had remaining maturities ranging from 12 to 17 months. None of our marketable securities changed from one fair value hierarchy to another during the year ended December 31, 2019.
4. Composition of Certain Balance Sheet Items
Inventory
Year Ended December 31, | |||||||
2019 | 2018 | ||||||
(in thousands) | |||||||
Raw materials | $ | $ | |||||
Work in progress | |||||||
Finished goods | |||||||
Total inventory | |||||||
Less strategic inventory classified as non-current | ( | ) | ( | ) | |||
Total inventory classified as current | $ | $ | |||||
F-14
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, Continued
Because we rely on a single manufacturer for the active pharmaceutical ingredient (“API”) for Korlym, we have purchased and hold significant quantities of API. We classify inventory we do not expect to sell within 12 months of the balance sheet date as “Strategic Inventory,” a long-term asset.
Property and Equipment
Year Ended December 31, | |||||||
2019 | 2018 | ||||||
(in thousands) | |||||||
Furniture and equipment | $ | $ | |||||
Software | |||||||
Leasehold improvements | |||||||
Less accumulated depreciation | ( | ) | ( | ) | |||
$ | $ | ||||||
Accrued and other liabilities
Year Ended December 31, | |||||||
2019 | 2018 | ||||||
(in thousands) | |||||||
Government rebates | $ | $ | |||||
Accrued compensation | |||||||
Legal fees | |||||||
Income taxes payable | |||||||
Accrued selling and marketing costs | |||||||
Professional fees | |||||||
Accrued manufacturing costs | |||||||
Other | |||||||
Total accrued and other liabilities | $ | $ | |||||
Other assets
5. Leases
We lease our office facilities in Menlo Park, California. On January 1, 2019, we recognized a right-of-use asset and a corresponding lease liability of $1.9 million. Effective October 1, 2019, we amended the lease to extend its term from March 31, 2020 through March 31, 2022 and to additional space beginning April 1, 2020. As a result of this amendment, we recognized an additional right-of-use asset and corresponding lease liability of $3.0 million. The right-of-use asset and lease liability recognized equals the present value of remaining payments due under our amended lease.
As our operating lease does not provide an implicit interest rate, we calculated the present value of remaining lease payments using a discount rate equal to the interest rate we would pay on a loan with monthly payments and a term equal to the monthly payments and remaining term of our lease. We recognize operating lease payments as expenses using the straight-line method over the term of the lease.
Operating lease expense for the year ended December 31, 2019 was approximately $1.5 million. Rent expense for the years ended December 31, 2018 and 2017 was $1.3 million and $1.1 million, respectively.
F-15
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, Continued
For any future operating lease transactions, we will recognize operating lease right-of-use assets and liabilities equal to the present value of the expected lease payments at the lease commencement date.
Our right-of-use assets and related lease liabilities were as follows:
Year Ended December 31, 2019 | |||
(in thousands) | |||
Cash paid for operating lease liabilities | $ | ||
Right-of-use assets obtained in connection with operating lease obligations | $ | ||
Remaining lease term (years) | |||
Discount rate | % | ||
As of December 31, 2019, future minimum lease payments under non-cancelable operating leases were as follows:
2020 | $ | ||
2021 | |||
2022 | |||
Less imputed interest | ( | ) | |
Total lease liability | $ | ||
6. Related Party Transactions
7. Preferred Stock and Stockholders’ Equity
Preferred Stock
Our Board of Directors is authorized, subject to any limitations prescribed by law, without stockholder approval, to issue up to an aggregate of 10,000,000 shares of preferred stock at $0.001 par value in one or more series and to fix the rights, preferences, privileges and restrictions granted to or imposed upon the preferred stock, including voting rights, dividend rights, conversion rights, redemption privileges and liquidation preferences. The rights of the holders of common stock will be subject to the rights of holders of any preferred stock that may be issued in the future. As of December 31, 2019 and 2018, we had no outstanding shares of preferred stock.
Common Stock
Significant stock transactions
On August 9, 2018, we announced a program to repurchase up to $100 million of our common stock (the “Stock Repurchase Program”). The terms of this program did not require us to acquire any shares and allowed for repurchases by a variety of methods, including in the open market, in block trades, through privately negotiated transactions, accelerated share repurchase transactions or any combination of such methods. The Stock Repurchase Program expired on June 30, 2019.
During the year ended December 31, 2019, we repurchased 2.8 million shares of common stock under the Stock Repurchase Program in open market transactions at a cost of $31.0 million (average price of $11.14 per share). In total, we repurchased 4.6 million shares under the Stock Repurchase Program at a cost of $54.6 million (an average price of $11.91 per share). We recorded repurchased shares as treasury stock on our consolidated balance sheet, at cost. We have not decided whether repurchased shares will be retired or sold.
During the year ended December 31, 2019, we issued 1.2 million shares as part of net-share settlements of cashless option exercises, of which 0.6 million shares were tendered to satisfy the related cost and statutory withholding requirements. We had no such transactions during the years ended December 31, 2018 and 2017.
F-16
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, Continued
We have never declared or paid any dividends.
Shares of common stock reserved for future issuance as of December 31, 2019 are as follows:
Common stock: | (in thousands) | |
Exercise of outstanding options | ||
Shares available for grant under stock option plans | ||
On February 7, 2020, our Board of Directors authorized an additional increase of 4.6 million shares in the number of shares available under the 2012 Equity Incentive Plan (the 2012 Plan), which was equivalent to 4 % of the shares of our common stock outstanding at December 31, 2019.
Stock Option Plans
We have two active stock option plans at December 31, 2019 – the 2004 Equity Incentive Plan (the 2004 Plan) and the 2012 Plan.
In 2004, our board of directors and stockholders approved the 2004 Plan, which became effective upon the completion of our initial public offering (IPO). Under the 2004 Plan, options, stock purchase and stock appreciation rights and restricted stock awards can be issued to our employees, officers, directors and consultants. The 2004 Plan provided that the exercise price for incentive stock options will be no less than 100 % of the fair value of the Company’s common stock, as of the date of grant. Options granted under the 2004 Plan vest over periods ranging from one year to five years . The vesting period of the options is generally equivalent to the requisite service period.
In 2012, our board of directors and stockholders approved the 2012 Plan. As of the effective date of the 2012 Plan, 5.3 million shares that remained available for issuance of new grants under the 2004 Plan were transferred to the 2012 Plan. After that date, no additional options were or will be issued under the 2004 Plan. Vested options under the 2004 Plan that are not exercised within the remaining contractual life and any options under the 2004 Plan that do not vest because of terminations after the effective date of the 2012 Plan will be added to the pool of shares available for future grants under the 2012 Plan.
Under the 2012 Plan, we can issue options, stock purchase and stock appreciation rights and restricted stock awards to our employees, officers, directors and consultants. The 2012 Plan provides that the exercise price for incentive stock options will be no less than 100 percent of the fair value of our common stock as of the date of grant. Options granted under the 2012 Plan are expected to vest over periods ranging from one to four years . We assume the vesting period of the options that we grant under the 2012 Plan to be equal to the option grantee’s period of service.
Upon exercise of options, new shares are issued.
F-17
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, Continued
Option activity during 2017, 2018 and 2019
The following table summarizes all activity under the 2004 Plan and the 2012 Plan:
Outstanding Options | |||||||||||||||
Shares Available For Future Grant | Options Shares Subject to Options Outstanding | Weighted- Average Exercise Price | Weighted- Average Remaining Contractual Life | Aggregate Intrinsic Value | |||||||||||
(in thousands) | (in thousands) | (in years) | (in thousands) | ||||||||||||
Balance at December 31, 2016 | $ | ||||||||||||||
Increase in shares authorized for grant | — | ||||||||||||||
Shares granted | ( | ) | $ | ||||||||||||
Shares exercised | — | ( | ) | $ | |||||||||||
Shares canceled and forfeited | ( | ) | $ | ||||||||||||
Balance at December 31, 2017 | $ | ||||||||||||||
Increase in shares authorized for grant | — | ||||||||||||||
Shares granted | ( | ) | $ | ||||||||||||
Shares exercised | — | ( | ) | $ | |||||||||||
Shares canceled and forfeited | ( | ) | $ | ||||||||||||
Balance at December 31, 2018 | $ | ||||||||||||||
Increase in shares authorized for grant | |||||||||||||||
Shares granted | ( | ) | $ | ||||||||||||
Shares exercised | — | ( | ) | $ | |||||||||||
Shares canceled and forfeited | ( | ) | $ | ||||||||||||
Balance at December 31, 2019 | $ | 6.51 | $ | ||||||||||||
Options exercisable at December 31, 2019 | $ | $ | |||||||||||||
Options fully vested and expected to vest at December 31, 2019 | $ | $ | |||||||||||||
The total intrinsic value of options exercised during the years ended December 31, 2019, 2018 and 2017 was $26.6 million, $26.6 million and $22.4 million, respectively, based on the difference between the closing price of our common stock on the date of exercise of the options and the exercise price.
The total grant date fair value of options to employees and directors that vested during the years ended December 31, 2019, 2018 and 2017 was $30.2 million, $22.6 million and $12.3 million, respectively.
F-18
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, Continued
The following is a summary of options outstanding and options exercisable at December 31, 2019.
Options Outstanding | Options Exercisable | |||||||||||||||||||||||||||||||
Exercise Prices of Options | Number of Shares | Weighted- Average Remaining Contractual Life | Weighted- Average Exercise Price | Aggregate Intrinsic Value | Number of Shares | Weighted- Average Exercise Price | Aggregate Intrinsic Value | |||||||||||||||||||||||||
(in thousands) | (in years) | (in thousands) | (in thousands) | (in thousands) | ||||||||||||||||||||||||||||
$ | — | $ | $ | $ | $ | $ | ||||||||||||||||||||||||||
$ | — | $ | $ | $ | ||||||||||||||||||||||||||||
$ | — | $ | $ | $ | ||||||||||||||||||||||||||||
$ | — | $ | $ | $ | ||||||||||||||||||||||||||||
$ | $ | $ | $ | |||||||||||||||||||||||||||||
The aggregate intrinsic value in the table above represents the total pre-tax intrinsic value that option holders would have received had all option holders exercised their options on December 31, 2019. The aggregate intrinsic value is the difference between our closing stock price on December 31, 2019 and the exercise price, multiplied by the number of options with exercise prices less than the closing stock price on that date.
Stock-Based Compensation related to Employee and Director Options
Assumptions used in determining fair value-based measurements for options to employees and directors
The following table summarizes the weighted-average assumptions and resultant fair value-based measurements for options granted to employees and directors.
Year Ended December 31, | |||||
2019 | 2018 | 2017 | |||
Weighted-average assumptions for stock options granted: | |||||
Risk-free interest rate | |||||
Expected term | |||||
Expected volatility of stock price | |||||
Dividend rate | |||||
Weighted-average grant date fair value-based measurement | $ | $ | $ | ||
The expected term of options reflected in the table above has been based on a formula that considers the expected service period and expected post-vesting termination behavior depending on whether the option holder is an employee, officer or director.
The expected volatility of our stock used in determining the fair value-based measurement of option grants to employees, officers and directors is based on the volatility of our stock price. The volatility is based on historical data of the price for our common stock for periods of time equal to the expected term of these grants.
We calculate employee stock-based compensation expense using the number of options we expect to vest, based on our estimate of the option grantees’ average length of employment, and reduced by our estimate of option forfeitures. ASC 718 requires us to estimate forfeitures at the time of option grant and revise this estimate in subsequent periods if actual forfeitures differ from our estimates.
Summary of compensation expense related to options to employees and directors
We recognized compensation expense of $29.2 million, $23.8 million and $13.4 million related to options to employees and directors during the years ended December 31, 2019, 2018 and 2017, respectively.
As of December 31, 2019, we had $55.0 million of unrecognized compensation expense for employee and director options outstanding as of that date, which had a weighted-average remaining vesting period of 2.48 years.
F-19
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, Continued
Stock Options to Non-Employees
We expense stock-based compensation related to service-based option grants to non-employees on a straight-line basis over the vesting period of the options, which approximates the period over which the related services are rendered, based on the options’ value as calculated by the Black-Scholes option pricing model. In performing this calculation we use the same assumptions as when determining the value of options granted to employees and directors, except that we use the remaining contractual term of the non-employee’s service as the options’ expected term and we recalculate the options’ value each quarter, based on the then current price of our common stock.
We recorded charges to expense for non-employee stock options of $0.2 million, zero and approximately zero for the years ended December 31, 2019, 2018 and 2017, respectively.
As of December 31, 2019, there were no awards outstanding to non-employees.
Summary of Stock-based Compensation Expense
The following table presents a summary of non-cash stock-based compensation by financial statement classification.
Year Ended December 31, | |||||||||||
2019 | 2018 | 2017 | |||||||||
(in thousands) | |||||||||||
Stock-based compensation capitalized in inventory | $ | $ | $ | ||||||||
Cost of sales | |||||||||||
Research and development | |||||||||||
Selling, general and administrative | |||||||||||
Total stock-based compensation | $ | $ | $ | ||||||||
8. Net Income Per Share
We compute basic and diluted net income per share by dividing our net income by the weighted-average number of common shares outstanding during the period. We used the treasury stock method to determine the number of dilutive shares of common stock resulting from the potential exercise of stock options. The statements of consolidated comprehensive income show the computation of net income per share for each period, including the number of weighted-average shares outstanding.
The following table shows the computation of net income per share for each period:
Year Ended December 31, | |||||||||||
2019 | 2018 | 2017 | |||||||||
(in thousands, except per share data) | |||||||||||
Numerator: | |||||||||||
Net income | $ | $ | $ | ||||||||
Denominator: | |||||||||||
Weighted-average shares used to compute basic net income per share | |||||||||||
Dilutive effect of employee stock options | |||||||||||
Weighted-average shares used to compute diluted net income per share | |||||||||||
Net income per share | |||||||||||
Basic | $ | $ | $ | ||||||||
Diluted | $ | $ | $ | ||||||||
As of December 31, 2019, 2018, and 2017 we had 23.6 million, 22.8 million, and 20.5 million stock options outstanding, respectively.
F-20
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, Continued
Because including them would have reduced dilution, we excluded from the computation of diluted net income per share, on a weighted-average basis 9.9 million, 5.0 million and 1.1 million stock options outstanding during the years ended December 31, 2019, 2018 and 2017, respectively,
9. Income Taxes
The domestic and foreign components of income before income taxes were as follows (in thousands):
Year Ended December 31, | |||||||||||
2019 | 2018 | 2017 | |||||||||
(in thousands) | |||||||||||
Domestic | $ | $ | $ | ||||||||
Foreign | |||||||||||
Income before income taxes | $ | $ | $ | ||||||||
The income tax expense (benefit) for the year ended December 31, 2019, 2018 and 2017 consisted of the following:
Year Ended December 31, | |||||||||||
2019 | 2018 | 2017 | |||||||||
(in thousands) | |||||||||||
U.S. federal taxes: | |||||||||||
Current | $ | $ | $ | ||||||||
Deferred | ( | ) | |||||||||
Total U.S. federal taxes | ( | ) | |||||||||
State taxes: | |||||||||||
Current | |||||||||||
Deferred | ( | ) | ( | ) | |||||||
Total state taxes | ( | ) | |||||||||
Total | $ | $ | $ | ( | ) | ||||||
The income tax benefit for the year ended December 31, 2017 resulted primarily from the partial release of our valuation allowance.
F-21
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, Continued
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of our deferred tax assets are as follows:
Year Ended December 31, | |||||||
2019 | 2018 | ||||||
Deferred tax assets: | (in thousands) | ||||||
Federal and state net operating losses | $ | $ | |||||
Capitalized research and patent costs | |||||||
Research credits | |||||||
Stock-based compensation costs | |||||||
Operating lease liability | |||||||
Other | |||||||
Total deferred tax assets | |||||||
Valuation allowance | ( | ) | ( | ) | |||
Deferred tax liabilities | |||||||
Operating lease right-of-use asset | ( | ) | |||||
Total deferred tax liabilities | ( | ) | |||||
Net deferred tax assets | $ | $ | |||||
Realization of deferred tax assets is dependent upon future earnings, if any, the timing and amount of which are uncertain. Each quarter, we assess our ability to use our deferred tax assets to offset our expected federal and state taxable income based on the weight of all available evidence, including such factors as the history of recent earnings and expected future taxable income on a jurisdiction by jurisdiction basis.
In the fourth quarter of 2017, we determined that it was more likely than not that we would generate sufficient taxable income to utilize our federal and state deferred tax assets in every state except California. We therefore included in our balance sheet the net value of all our deferred tax assets except those applicable to California. We maintain a full valuation allowance in relation to California deferred tax assets as of December 31, 2019 because of the uncertainty regarding the realizability of these deferred tax assets. All tax years from Corcept’s inception remain open to examination by the Internal Revenue Service, the California Franchise Tax Board and other state taxing authorities.
The valuation allowance increased by $0.2 million for the year ended December 31, 2019, and decreased by $1.3 million and $116.9 million for the years ended December 31, 2018 and 2017, respectively. The significant decrease in the valuation allowance during 2017 was the result of our release of the entire valuation allowance previously established on our federal and non-California state deferred tax assets.
At December 31, 2019, we had net operating loss carryforwards available to offset any future taxable income that we may generate for federal income tax purposes of $7.7 million, which will begin to expire in the year 2033, California net operating loss carryforwards of $75.2 million, which will begin to expire in the year 2029, and net operating loss carryforwards from other states of $9.7 million, which will begin to expire in the year 2023 if not utilized.
At December 31, 2019, we also had federal research and development tax credits of $10.4 million and orphan drug tax credits of $14.6 million, respectively and California research and development credits of $7.5 million. The federal tax credits will begin to expire in the years 2030 through 2039 and the California research credits have no expiration date.
Utilization of our net operating losses and tax credit carryforwards may be subject to substantial annual limitation due to the ownership change limitations provided by the Internal Revenue Code and similar state provisions. Such limitations could result in the expiration of the net operating losses and tax credit carryforwards before utilization.
F-22
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, Continued
The following table presents a reconciliation from the statutory federal income tax rate to the effective rate.
Year Ended December 31, | |||||||||||
2019 | 2018 | 2017 | |||||||||
(in thousands) | |||||||||||
U.S. federal taxes at statutory rate | $ | $ | $ | ||||||||
Changes in valuation allowance | ( | ) | |||||||||
Federal tax rate change impact to change in valuation allowance | |||||||||||
R&D and other credits | ( | ) | ( | ) | ( | ) | |||||
State income taxes | ( | ) | |||||||||
Non-deductible compensation | |||||||||||
Stock-based compensation | ( | ) | ( | ) | ( | ) | |||||
Other | |||||||||||
Total | $ | $ | $ | ( | ) | ||||||
We maintain liabilities for uncertain tax positions. The measurement of these liabilities involves considerable judgment and estimation and are continuously monitored by management based on the best information available, including changes in tax regulations, the outcome of relevant court cases, and other pertinent information.
The aggregate annual changes in the balance of gross unrecognized tax benefits are as follows (in thousands):
Year Ended December 31, | |||||||||||
2019 | 2018 | 2017 | |||||||||
Beginning Balance | $ | $ | $ | ||||||||
Increase in tax positions for prior years | |||||||||||
Decrease in tax positions for prior years | ( | ) | |||||||||
Increase in tax positions for current year | |||||||||||
Decrease in tax positions for current year | |||||||||||
Ending Balance | $ | $ | $ | ||||||||
As of December 31, 2019, 2018 and 2017, the total amount of unrecognized tax benefits was approximately $6.0 million, $4.8 million and $4.1 million, respectively. A valuation allowance is maintained on the tax benefits related to California deferred tax assets and if these tax benefits were recognized it would not impact the effective tax rate. We had no or immaterial amounts of accrued interest and no accrued penalties related to unrecognized tax benefits as of December 31, 2019, 2018 and 2017. We do not expect our unrecognized tax benefits to change materially over the next 12 months.
While we believe we have adequately provided for all tax positions, amounts asserted by tax authorities could be greater or less than the recorded position. Accordingly, our provisions on federal and state tax-related matters to be recorded in the future may change as revised estimates are made or the underlying matters are settled or otherwise resolved.
10. Commitments and contingencies
We have entered into a number of agreements to purchase API for the manufacturing of relacorilant, miricorilant and exicorilant. We have also entered into a number of agreements to perform clinical studies on miricorilant and CORT113176. See the discussion in Note 2, Significant Agreements, for further discussion regarding the commitments under these agreements.
In the ordinary course of business, we may be subject to legal claims and regulatory actions that could have a material adverse effect on our business or financial position. We assess our potential liability in such situations by analyzing the possible
F-23
CORCEPT THERAPEUTICS INCORPORATED
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, Continued
outcomes of various litigation, regulatory and settlement strategies. If we determine a loss is probable and its amount can be reasonably estimated, we accrue an amount equal to the estimated loss.
In August 2017, we terminated our pharmaceutical services agreement with our exclusive specialty pharmacy, Dohmen Life Science Services ("Dohmen") for material breach. In August 2017, Dohmen filed a complaint in the Court of Chancery of the State of Delaware against us alleging unlawful termination and breach of contract and requesting declaratory relief and damages. We filed a complaint against Dohmen in the Superior Court of the State of Delaware and a motion to dismiss the Dohmen complaint against us. In November 2017, we answered Dohmen’s complaint in the Court of Chancery of the State of Delaware and asserted counterclaims against Dohmen.
Dohmen refused to transfer to us the cash it collected from $12.9 million in Korlym® receivables, despite its obligation to do so. As of December 31, 2017, the total amount of these receivables had been included in “Other receivable” on our consolidated balance sheet.
In January 2018, we entered into a settlement agreement with Dohmen and mutual release of any and all claims that may have existed between the parties as of that date, pursuant to which Dohmen agreed to deliver to us the cash it had collected from the sale of Korlym on our behalf. The total amount delivered by Dohmen under the settlement agreement was the $12.9 million of Korlym® receivables described above.
11. Quarterly Financial Data (Unaudited)
The following table is in thousands, except per share amounts:
Quarter Ended | March 31 | June 30 | September 30 | December 31 | |||||||||||
2019 | |||||||||||||||
Product revenue, net | $ | $ | $ | $ | |||||||||||
Gross profit on product revenue | |||||||||||||||
Net income | |||||||||||||||
Basic net income per share | $ | $ | $ | $ | |||||||||||
Diluted net income per share | $ | $ | $ | $ | |||||||||||
2018 | |||||||||||||||
Product revenue, net | $ | $ | $ | $ | |||||||||||
Gross profit on product revenue | |||||||||||||||
Net income | |||||||||||||||
Basic net income per share | $ | $ | $ | $ | |||||||||||
Diluted net income per share | $ | $ | $ | $ | |||||||||||
F-24