UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
_______________
Form 10-K
_______________
|
|
(Mark One) |
|
☑ |
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the Fiscal Year Ended December 31, 2019
or
|
☐ |
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from _____ to _____
Commission File Number: 000-29959
Cassava Sciences, Inc.
(Exact name of registrant as specified in its charter)
|
Delaware |
91-1911336 |
|
(State or other jurisdiction of |
(I.R.S. Employer |
|
incorporation or organization) |
Identification Number) |
7801 N. Capital of Texas Highway, Suite 260, Austin, TX 78731
(512) 501-2444
(Address, including zip code, of registrant's principal executive offices and
telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
|
Title of each class |
|
Trading Symbol(s) |
|
Name of each exchange on which registered |
|
Common Stock, $0.001 par value |
|
SAVA |
|
NASDAQ Capital Market |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☑
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☑
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☑ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☑ No ☐.
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act. (Check one):
|
Large accelerated filer ☐ |
Accelerated filer ☐ |
|
Non-accelerated filer ☑ |
Smaller reporting company ☑ |
|
|
Emerging growth company ☐ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ☐ No ☑
The aggregate market value of the voting and non-voting common equity held by non-affiliates was $19,381,188 computed by reference to the last sales price of $1.21 as reported on the Nasdaq Capital Market, as of the last business day of the Registrant's most recently completed second fiscal quarter, June 28, 2019. The number of shares outstanding of the Registrant's common stock on March 19, 2020 was 24,729,902.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the Registrant's Proxy Statement for its 2020 Annual Meeting of Stockholders (the Proxy Statement), to be filed with the U.S. Securities and Exchange Commission, are incorporated by reference to Part III of this Form 10-K Report.
1
CASSAVA SCIENCES, INC.
FORM 10-K
INDEX
|
|
|
|
|
|
|
Page |
|
PART I |
||
|
Item 1. |
4 |
|
|
Item 1A. |
24 |
|
|
Item 1B. |
61 |
|
|
Item 2. |
61 |
|
|
Item 3. |
61 |
|
|
Item 4. |
61 |
|
|
|
|
|
|
PART II |
||
|
Item 5. |
62 |
|
|
Item 6. |
62 |
|
|
Item 7. |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
63 |
|
Item 7A. |
69 |
|
|
Item 8. |
70 |
|
|
Item 9. |
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
86 |
|
Item 9A. |
86 |
|
|
Item 9B. |
87 |
|
|
|
|
|
|
PART III |
||
|
Item 10. |
87 |
|
|
Item 11. |
87 |
|
|
Item 12. |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
87 |
|
Item 13. |
Certain Relationships and Related Transactions and Director Independence |
88 |
|
Item 14. |
88 |
|
|
|
|
|
|
PART IV |
||
|
Item 15. |
89 |
|
|
Item 16. |
90 |
|
2
PART I
FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains forward-looking statements within the meaning of the Private Securities Reform Act of 1995. All statements other than statements of historical facts contained in this Annual Report are forward-looking statements. We intend that such statements be protected by the safe harbor created thereby. Forward-looking statements relate to expectations, beliefs, projections, future plans and strategies, anticipated events or trends and similar expressions concerning matters that are not historical facts. In some cases, you can identify forward-looking statements by terms such as “anticipate,” “believe,” “could,” “estimate,” “expect,” “intend,” “may,” “plan,” “potential,” “should,” “will” and “would” or the negatives of these terms or other comparable terminology.
The forward-looking statements are based on our beliefs, assumptions and expectations of our future performance, taking into account all information currently available to us. Forward-looking statements involve risks and uncertainties and our actual results and the timing of events may differ significantly from the results discussed in the forward-looking statements. Such forward-looking statements and our business are subject to numerous risks and uncertainties that you should consider before investing in our Company. These risks are described more fully in the section titled “Risk Factors.” Accordingly, you should not rely upon forward-looking statements as predictions of future events. Examples of such forward-looking statements include, but are not limited to statements about:
|
· |
Our ability to initiate, conduct or analyze studies with PTI-125 or SavaDx (formerly referred to as PTI-125Dx), our lead product candidates targeted at Alzheimer’s disease and other neurodegenerative diseases; |
|
· |
our estimated timeline for announcing top-line results of a Phase 2b study of PTI-125; |
|
· |
our estimated timeline for testing clinical samples with SavaDx, our investigational blood-based diagnostic; |
|
· |
our intention to conduct additional clinical studies of PTI-125 or SavaDx, the anticipated scope of such studies and our estimated timeline for doing so; |
|
· |
the beneficial characteristics, safety, efficacy, and therapeutic effects of our product candidates, such as PTI-125 or SavaDx; |
|
· |
the utility of protection, or the sufficiency, of our intellectual property; |
|
· |
our potential competitors or competitive products; |
|
· |
expected future sources of revenue and capital and increasing cash needs; |
|
· |
our continued reliance on third parties to conduct additional clinical studies of our product candidates, and for the manufacture of our product candidates; |
|
· |
expectations regarding trade secrets, technological innovations, licensing agreements and outsourcing of certain business functions; |
|
· |
our expenses increasing or fluctuations in our financial or operating results; |
|
· |
our operating losses and anticipated operating and capital expenditures; |
|
· |
expectations regarding the issuance of shares of common stock to employees pursuant to equity compensation awards, net of employment taxes; |
|
· |
our ability to maintain compliance with the ongoing listing requirements for the Nasdaq Capital Market; |
|
· |
the development and maintenance of our internal systems and infrastructure; |
3
|
· |
our need to hire additional personnel and our ability to attract and retain such personnel; |
|
· |
existing regulations and regulatory developments in the United States and other jurisdictions; |
|
· |
the sufficiency of our current resources to continue to fund our operations; |
|
· |
the accuracy of our estimates regarding expenses, capital requirements, and needs for additional financing; and |
|
· |
assumptions and estimates used for our disclosures regarding stock-based compensation. |
We cannot assure you that we will realize the results or developments we expect or anticipate or, even if substantially realized, that they will result in the consequences or affect us or our operations in the way we expect. The forward-looking statements included in this Annual Report on Form 10-K are made only as of the date hereof. We undertake no obligation to publicly update or revise any forward-looking statement as a result of new information, future events or otherwise, except as required by law.
In addition, statements that “we believe” and similar statements reflect our beliefs and opinions on the relevant subject. These statements are based upon information available to us as of the date of this report, and while we believe such information forms a reasonable basis for such statements, such information may be limited or incomplete, and our statements should not be read to indicate that we have conducted an exhaustive inquiry into, or review of, all potentially available relevant information. These statements are inherently uncertain and you are cautioned not to unduly rely upon these statements.
Our research programs in neurodegeneration benefits from longstanding scientific and financial support from the National Institutes of Health (NIH). The contents of this Annual Report are solely our responsibility and do not necessarily represent any official views of NIH.
Overview
Cassava Sciences, Inc. is a clinical stage biotechnology company. Our mission is to detect and treat neurodegenerative diseases, such as Alzheimer’s disease. Our novel science is based on stabilizing – but not removing – a critical protein in the brain.
Over the past 10 years, we have combined state-of-the-art technology with new insights in neurobiology to develop novel solutions for Alzheimer’s disease and other neurodegenerative diseases. Our strategy is to leverage our unique scientific/clinical platform to develop a first-in-class program for treating neurodegenerative diseases, such as Alzheimer’s.
We currently have two clinical-stage biopharmaceutical assets under development:
|
· |
our lead therapeutic product candidate, called PTI-125, for the treatment of Alzheimer’s disease; and |
|
· |
our lead investigational diagnostic product candidate, called SavaDx, to detect Alzheimer’s disease from a small sample of blood, possibly years before the overt appearance of clinical symptoms. |
Our scientific approach for the treatment of Alzheimer’s disease seeks to simultaneously improve both neurodegeneration and neuroinflammation. We believe our ability to improve multiple vital functions in the brain represents a new, different and crucial approach to address Alzheimer’s disease.
Our lead therapeutic product candidate, PTI-125, is a proprietary small molecule (oral) drug. PTI-125 targets an altered form of a protein called filamin A (FLNA) in the Alzheimer’s brain. Published studies have demonstrated that the altered form of FLNA causes neuronal dysfunction, neuronal degeneration and neuroinflammation.
We believe PTI-125 improves brain health by reverting altered FLNA back to its native, healthy confirmation, thus countering the downstream toxic effects of altered FLNA. We have generated and published experimental and clinical
4
evidence of improved brain health with PTI-125. Importantly, PTI-125 is not dependent on clearing amyloid from the brain. Since PTI-125 has a unique mechanism of action, we believe its potential therapeutic effects may be additive or synergistic with that of other therapeutic candidates aiming to treat neurodegeneration.
PTI-125 has demonstrated a multitude of beneficial effects in animal models of disease, including normalizing neurotransmission, decreasing neuroinflammation, suppressing neurodegeneration, and restoring memory and cognition.
In 2019, we completed a small, first-in-human, clinical-proof-of-concept, open-label Phase 2a study of PTI-125 in the U.S., with substantial support from the National Institute on Aging (NIA), a division of the NIH. Treatment with PTI-125 for 28 days significantly improved key biomarkers of Alzheimer’s pathology, neurodegeneration and neuroinflammation (p<0.001). Biomarkers effects were seen in all patients in both cerebrospinal fluid (CSF) and plasma. To our knowledge, no other drug candidate has improved an entire panel of biomarkers of in patients with Alzheimer’s disease.
A confirmatory, randomized, placebo-controlled, multi-center Phase 2b study of PTI-125 in Alzheimer’s disease is on-going as of March 2020. We expect to announce Phase 2b results in approximately mid-2020.
Our diagnostic effort, called SavaDx, is an early-stage program focused on detecting Alzheimer’s disease from a small sample of blood, possibly years before the overt appearance of clinical symptoms. We are developing SavaDx as a fast, accurate and quantitative blood-based investigational biomarker/diagnostic to detect and monitor Alzheimer's disease. The goal is to make the detection of Alzheimer’s disease as simple as getting a blood test.
Alzheimer’s disease is a progressive neurodegenerative disorder that affects cognition, function and behavior. An estimated 5.8 million Americans are living with Alzheimer’s disease in 2020, according to the Alzheimer’s Association, a non-profit organization. There are no disease-modifying drug therapies to treat the disease.
PTI-125 and SavaDx were both discovered and designed in-house and were characterized by our academic collaborators during research activities that were conducted from approximately 2008 to date. We own exclusive, worldwide rights to these drug assets and related technologies, without royalty obligations to any third party. Our patent protection in this area currently runs through 2034.
Our Scientific Approach is Different.
Over the last ten years, we have developed a new and promising scientific approach for the treatment and diagnosis of neurodegenerative diseases, such as Alzheimer’s disease. Importantly, we do not seek to clear amyloid out of the brain. Rather, we seek to stabilize a critical protein in the brain that has many downstream effects.
Our scientific approach is to treat neurodegeneration by targeting an altered form of a scaffolding protein called FLNA. Through years of basic research, we and our academic collaborators identified FLNA as a structurally altered protein in the Alzheimer’s brain. We have shown that the altered form of FLNA is pervasive in the Alzheimer’s brain and undetectable in healthy control brains.
Using scientific insight and advanced techniques in molecular biochemistry, bioinformatics and imaging, we have elucidated this protein dysfunction. Through this work, we have produced experimental evidence that altered FLNA plays a critical role in Alzheimer’s disease. We engineered a family of high-affinity, small molecules to target this structurally altered protein and restore its normal shape and function. This family of small molecules, including our lead therapeutic candidate, PTI-125, was designed in-house and characterized by our academic collaborators.
Our lead therapeutic product candidate, PTI-125, is a small molecule (oral) drug with a novel mechanism of action. The target of PTI-125 is altered FLNA, the brain protein we seek to stabilize. Importantly, since PTI-125 has a unique mechanism of action, we believe its potential therapeutic effects may be additive or synergistic with that of other therapeutic candidates aiming to treat neurodegeneration.
Given the biopharmaceutical industry’s challenging track record in Alzheimer’s research, we believe there is an urgent need to consider more recent and innovative approaches to combat this disease. We believe our scientific approach may broaden the range of possible treatment approaches for this complex disease.
5
Our science is based on stabilizing a critical protein in the brain.
Proteins are essential for cell function because they participate in virtually every biological process. If protein function is impaired, the health consequences can be devastating. Technological advances in medicine and improvements in lifestyle are making our lives longer. But with age, genetic mutations and other factors conspire against healthy cells, resulting in altered proteins. Sometimes a cell can rid itself of altered proteins. However, when disease changes the shape and function of critical proteins, multiple downstream processes are impaired. There are many clinical conditions in which proteins become structurally altered and impair the normal function of cells, tissues and organs, leading to disease. Conversely, restoring altered proteins back to health –called proteostasis – is a well-accepted therapeutic strategy in clinical medicine.
For over 100 years, scientists have ascribed various neurodegenerative diseases to proteins that misfold and are rendered pathological. In Alzheimer’s disease, certain proteins, such as amyloid and tau, lose their normal shape and function. Such misfolded proteins can breakdown or aggregate in clumps and form plaque in the brain. Destruction of neuronal synapses, accelerated nerve cell death, and dysfunction of the brain support cells, are all widely believed to be direct consequences of misfolded proteins.
FLNA is a scaffolding protein widely found throughout the body. A healthy scaffolding protein brings multiple proteins together, initiating their interaction. However, an altered form of FLNA protein is found in the Alzheimer’s brain. Our experimental evidence shows that altered FLNA protein contribute to Alzheimer’s disease by disrupting the normal function of neurons, leading to neurodegeneration and brain inflammation. Our product candidate, PTI-125, aims to counter the altered and toxic form of FLNA in the brain, thus restoring the normal function of this critical protein. Our novel science is based on stabilizing – but not removing – a critical protein in the brain.
One drug, multiple effects.
Our lead therapeutic candidate, PTI-125, binds to altered FLNA with very high (femtomolar) affinity. This drug effect restores the normal shape of FLNA and the normal function of three key brain receptors: the alpha-7 nicotinic acetylcholine receptor; the N-methyl-D-aspartate (NMDA) receptor; and the insulin receptor. These receptors have pivotal roles in brain cell survival, cognition and memory.
In animal models, treatment with PTI-125 resulted in dramatic improvements in brain health, such as reduced amyloid and tau deposits, improved receptor signaling and improved learning and memory. In addition, PTI-125 has another beneficial treatment effect of significantly reducing inflammatory cytokines in the brain. In animal models of disease, treatment with PTI-125 greatly reduced levels of IL-6 and suppressed TNF-alpha and IL-1beta levels by 86% and 80%, respectively, illustrating a powerful anti-neuroinflammatory effect.
In 2019, a small, first-in-patient, clinical proof-of-concept Phase 2a study funded by NIH, showed that open-label treatment with PTI-125 for 28 days significantly improved key biomarkers of Alzheimer’s pathology, neuroinflammation and neurodegeneration (p<0.001). By restoring function to multiple receptors and exerting powerful anti-inflammatory effects, we believe our approach has potential to slow the progression of neurodegeneration in patients. Thus, we have designed PTI-125 to slow or, potentially, even reverse the deterioration of brain cells. We believe the ability to simultaneously improve many vital functions in the brain represents a new, different and crucial approach to combating neurodegeneration.
Our science is published in multiple peer-reviewed journals. In addition, our research has been supported by NIH under multiple research grant awards. Each grant was awarded following an in-depth, peer-reviewed evaluation of our approach for scientific and technical merit by a panel of outside experts in the field. Strong, long-term support from NIH has allowed us to advance our two product candidates for neurodegeneration, PTI-125 and SavaDx, into clinical development.
Overview of Alzheimer’s disease.
Alzheimer’s disease is a progressive neurodegenerative disorder that has debilitating effects on patients' cognition, memory and day-to-day functioning. Most cases of Alzheimer’s disease are age-related. Alzheimer’s disease has become markedly more common with the aging of the U.S. population. The prevalence of Alzheimer's disease is widely expected to increase over time, with 13.8 million people age 65 and older projected to have the disease by 2050 in the U.S., up from 5.6 million in 2019.
6
According to the non-profit Alzheimer's Association, the aggregate cost of care in 2019 for patients with Alzheimer's disease and other types of dementia in the U.S. was estimated to be $234 billion. The prevalence of Alzheimer’s disease is expected to nearly triple in the U.S. between now and 2050. If this occurs, there is potential for Alzheimer's disease to cause a major financial drain on the national economy.
Currently marketed drug therapies for Alzheimer’s disease have limited therapeutic effect.
There are no disease-modifying drug therapies to treat Alzheimer’s disease. The U.S. Food & Drug Administration (FDA) has not approved any new drugs for Alzheimer’s disease since 2003. Currently marketed drug therapies focus solely on treating symptoms, mostly in patients with mild-to-moderate Alzheimer's disease. At the time of diagnosis, patients are initiated on a class of drugs called cholinesterase inhibitors. The Alzheimer’s brain has low levels of a neurotransmitter called acetylcholine. Cholinesterase inhibitors prevent an enzyme in the brain, called acetylcholinesterase, from breaking down acetylcholine. Currently marketed cholinesterase inhibitors include donepezil (marketed by Eisai Co., Ltd. and Pfizer, Inc. as Aricept®), rivastigmine (marketed by Novartis AG as Exelon®) and galantamine (marketed by Janssen Pharmaceuticals, Inc. as Razadyne®). Cholinesterase inhibitors may benefit some patients for several months, after which the targeted brain receptors are desensitized, and drug efficacy is lost.
PTI-125 is our Proprietary Drug for the Treatment of Alzheimer’s Disease.
We have generated and published experimental evidence of improved brain health by restoring altered FLNA with PTI-125, our lead therapeutic product candidate. PTI-125 is a proprietary small molecule drug that represents an entirely new scientific approach to treat neurodegeneration. Published studies have demonstrated that PTI-125 targets an altered form of a protein called FLNA that is pervasive in the Alzheimer’s brain. Altered FLNA causes neuronal dysfunction, neuronal degeneration and neuroinflammation. We believe our drug candidate, PTI-125, improves brain health by reverting altered FLNA back to its native, healthy confirmation, thus countering downstream toxic effects of altered FLNA. Importantly, PTI-125 is not dependent on clearing amyloid from the brain. The following is a summary profile of PTI-125’s drug development program.
IND submission to FDA.
Over the past ten years, we successfully conducted basic research, in vitro studies and preclinical studies in support of an Investigational New Drug (IND) submission to the FDA for PTI-125, including requisite studies around safety pharmacology, toxicology, genotoxicity and bioanalytical methods. In 2017 we filed an IND with FDA for PTI-125.
Clinical safety of PTI-125 in a Phase 1 study.
Following FDA acceptance of our IND in 2017, we investigated the safety, dosing and pharmacokinetic profile of PTI-125 in healthy human volunteers. The design of our first-in-human Phase 1 study was based on regulatory feedback, clinical and scientific rationale and observations from previously conducted preclinical and in vitro studies.
In a Phase 1 study, PTI-125 was evaluated in 24 healthy human volunteers in a single site in the U.S. for safety, tolerability and pharmacokinetics. Study subjects were administered a single oral dose of 50, 100 or 200 mg of PTI-125. Drug was well-tolerated in all subjects. Importantly, PTI-125 showed no treatment-related adverse effects and no dose-limiting safety findings. Pharmacokinetic measurements demonstrated that PTI-125, a small molecule, was rapidly absorbed. Dose-proportionality was observed over the full dose range of 50 to 200 mg. Results of this Phase 1 study were presented at the 10th Annual International Conference on Clinical Studies on Alzheimer's Disease in 2017 (Figure 1).
7
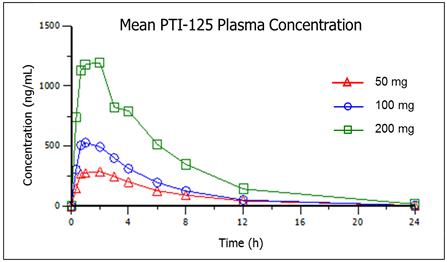
Figure 1. Phase 1 study results with PTI-125 in healthy volunteers demonstrated dose-proportional pharmacokinetics.
Given the absence of any observable dose-limiting effects in healthy adults in a Phase 1 study, a strong scientific rationale, and multiple peer-reviewed publications and research grant awards, we believe this program demonstrated favorable proof-of-principle for the development of PTI-125 in Alzheimer’s disease.
Phase 2 clinical development program for PTI-125.
In 2018, we mapped out, in collaboration with outside science advisors and medical experts, a strategic clinical plan to advance PTI-125 into a comprehensive Phase 2 clinical development program. The general objective of our Phase 2 program with PTI-125 is to gain an initial estimate of the safety, tolerability and biological activity of this drug candidate in patients with Alzheimer’s disease. We believe meaningful Phase 2 data may enable us to design and conduct a large-scale Phase 3 efficacy program for PTI-125 and may also enable us to seek one or more strategic collaborations with pharmaceutical or biotechnology companies.
Phase 2a Clinical Study
In 2019, we completed a first-in-patient, clinical proof-of-concept study of PTI-125 in the U.S. Our Phase 2a was an open-label, multi-center, safety and pharmacokinetic study of PTI-125. Thirteen (13) patients with mild-to-moderate Alzheimer’s disease, age 50-85, received 100 mg oral PTI-125 twice daily for 28 days. A diagnosis of Alzheimer’s disease was confirmed with Mini-Mental State Examination (MMSE) ≥ 16 and ≤ 24 and a cerebrospinal fluid (CSF) T-tau/Aβ42 ratio ≥ 0.30. Safety was assessed by ECGs, clinical labs, adverse event monitoring and physical examinations. CSF was drawn from patients before dosing started and again after 28 continuous days of dosing with PTI-125. CSF samples were then analyzed for biomarkers of Alzheimer’s pathology (T-tau, P-tau, Aβ42); neurodegeneration (NfL, neurogranin); and neuroinflammation (YKL-40, IL-6, IL-1β and TNFα). A consulting biostatistician conducted an independent analysis of the data set.
A key objective of our Phase 2a study was to measure levels of biomarkers in the brain. Key results of this study include (Figure 2):
|
· |
Total tau (T-tau) decreased 20% (p<0.001) |
|
· |
Phosphorylated tau (P-tau) decreased 34% (p<0.0001) |
|
· |
Neurofilament light chain (NfL), a marker for neurodegeneration, decreased 22% (p<0.0001) |
|
· |
Neurogranin, a marker for cognitive decline, decreased 32% (p<0.0001) |
|
· |
Neuroinflammatory marker YKL-40, an indicator of microglial activation, decreased 9% (p<0.0001) |
|
· |
Proinflammatory Interleukin 6 (IL-6) decreased 14% (p<0.0001) |
8
|
· |
Proinflammatory Interleukin 1 beta (IL-1β) decreased 11% (p<0.0001) |
|
· |
Proinflammatory Tumor Necrosis Factor alpha (TNFα) decreased 5% (p<0.001) |
|
· |
The ratio of CSF P-tau to Aβ42, a widely accepted biochemical value of Alzheimer’s disease, improved in all patients (p<0.001) |
Figure 2: PTI-125 treatment reduces levels of CSF biomarkers in patients with Alzheimer’s in a Phase 2a study.
|
|
|
|
|
|
|
|
|
|
Neurogranin* |
NfL* |
T-tau+ |
P-tau181* |
YKL40* |
IL-6* |
IL-1β* |
TNFα+ |
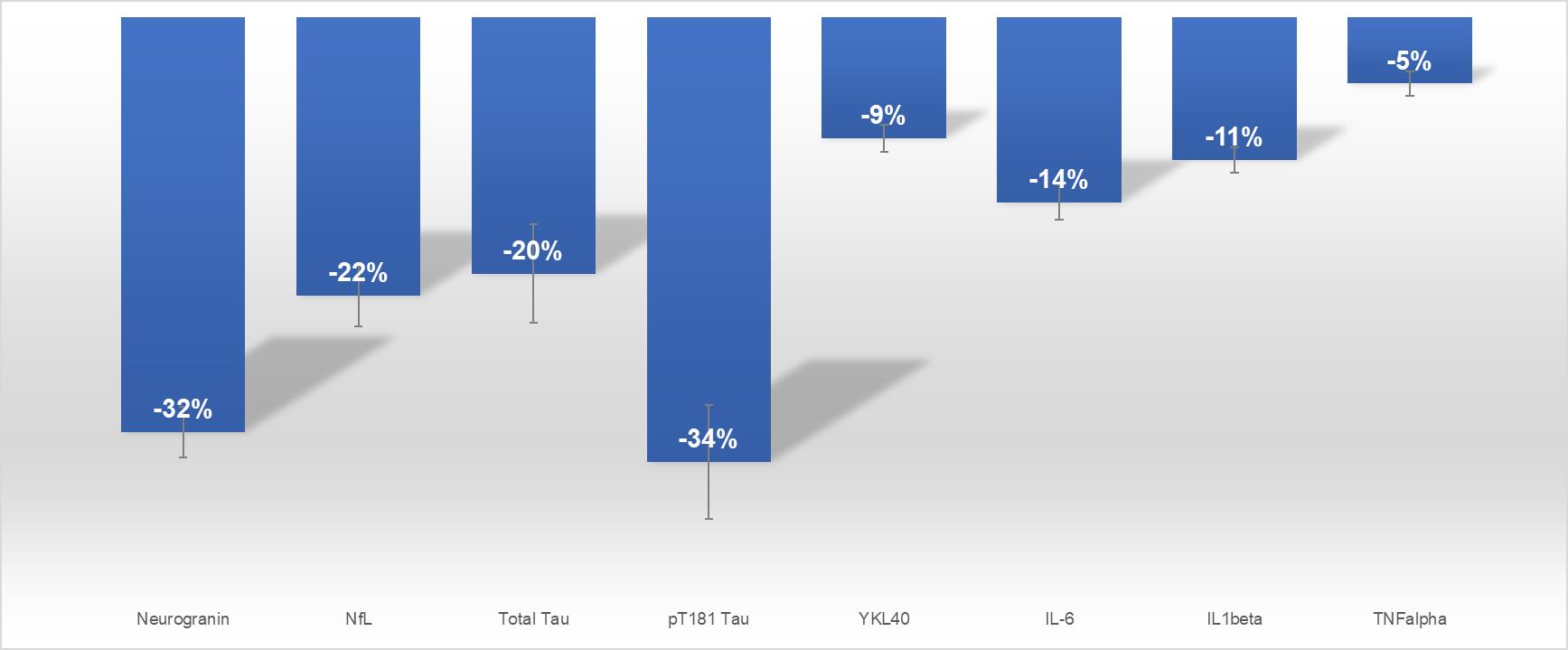
Figure 2. Percent change from baseline in CSF biomarkers measured by ELISA. Eight CSF biomarkers of disease in Alzheimer’s patients were significantly reduced with PTI-125 treatment. *p < 0.0001, +p < 0.001 in paired t test comparing Day 28 to pre-dose baseline.
In addition, visual line plots, which are also known as spaghetti plots, of each individual patient and each individual biomarker measurement show that all patients had a biomarker response to treatment with PTI-125 in our Phase 2a study (Figure 3).
Figure 3. Individual patient response to treatment with PTI-125 in a Phase 2a study. CSF data are plotted as pg/mL.
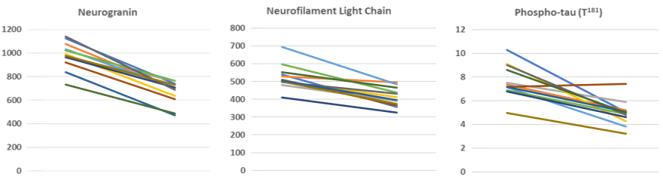
9
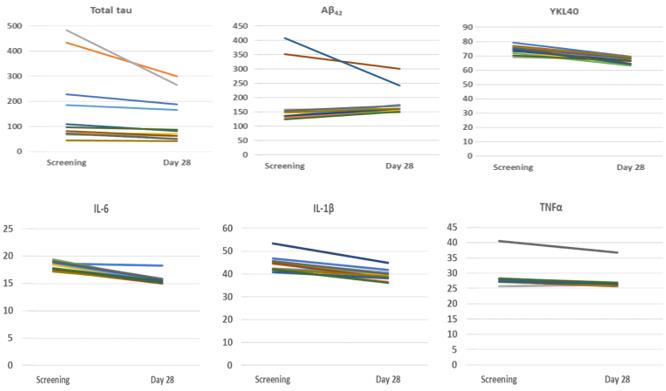
Figure 3. Each spaghetti plot shows change from baseline to Day 28 for each patient and each biomarker.
Consistent with over 10 years of basic research and pre-clinical data, we believe our Phase 2a study showed clinical evidence of PTI-125’s mechanism of action and drug-target engagement, including:
|
· |
Improvements in biomarkers of Alzheimer’s disease in CSF, plasma and lymphocytes; |
|
· |
Consistency across biomarker improvements in CSF, plasma, and lymphocytes; |
|
· |
Significant reductions (p<0.01) in both nitrated and phosphorylated forms of tau protein; |
|
· |
Evidence that each individual patient showed biomarker responses to PTI-125; |
|
· |
Evidence that PTI-125 reversed the shape of altered filamin A in lymphocytes; |
|
· |
Evidence that PTI-125 reduced levels of amyloid bound to alpha 7 nicotinic receptors in lymphocytes; |
|
· |
Early clinical validation of the drug target – altered filamin A – as a facilitator protein between amyloid beta and both neuroinflammation and tau pathology. |
To our knowledge, no competing drug candidate has demonstrated the ability to reduce an entire panel of biomarkers of disease in patients with Alzheimer’s disease. For this reason, CSF biomarker data generated in our Phase 2a study may not be directly comparable to clinical results generated by our competitors.
In December 2019, we presented data from our Phase 2a study at the 12th International Conference on Clinical Trials on Alzheimer’s Disease (CTAD), a conference for the medical and scientific community held in San Diego, CA. Our Phase 2a presentation was selected as a “Late Breaking Oral Communication” by CTAD 2019.
In February 2020, our Phase 2a study was published in The Journal of Prevention of Alzheimer’s Disease (JPAD), a technical journal for the research community. This peer-reviewed publication highlighted that biomarkers of Alzheimer’s
10
disease pathology (P-tau, total tau and Aβ42), neurodegeneration (NfL and neurogranin) and neuroinflammation (YKL-40, IL-6, IL-1β and TNFα) improved significantly after 28 days of treatment with PTI-125.
Cognition and function were not assessed in this small Phase 2a study. However, independent research has shown that high levels of CSF biomarkers of P-tau and total tau/Aβ42 ratio correlate with worse performance on a wide range of memory and attention tests. Conversely, lowering biomarkers of disease may benefit patients.
Phase 2b Clinical Study
In September 2019, we announced initiation of a Phase 2b confirmatory clinical study in Alzheimer’s patients, with funding provided by NIH. This Phase 2b clinical study is designed to evaluate safety, tolerability and drug effects of PTI-125 on biomarkers of Alzheimer’s disease. This is a blinded, randomized, placebo-controlled, multi-center, multi-dose research study in 64 patients with mild-to-moderate Alzheimer’s disease. Patients received PTI-125 100 mg, 50 mg, or matching placebo, twice daily for 28 continuous days. The primary endpoint is improvement in biomarkers of Alzheimer’s disease from baseline to Day 28.
In January 2020, we announced the completion of patient enrollment for our Phase 2b study. In February 2020, the last study participants were successfully dosed. In March 2020, the last study participants underwent final, routine follow-ups. No safety issues were found. Cerebrospinal fluid and plasma samples from study participants were then shipped to independent, third party labs for biomarker analysis. Study samples will be analyzed under blinded conditions, meaning no one will know whether a test sample came from a study participant who was on drug or placebo until the study is unblinded. Lab testing, statistical analysis, data analytics and interpretation of results are expected to run through approximately May 2020. We expect to announce Phase 2b results approximately in approximately mid-2020.
Open-label Extension Clinical Study
On March 25, 2020, we announced the initiation of an open-label extension study to evaluate PTI-125 in patients with Alzheimer’s disease. This open-label, multi-center, extension study will monitor the long-term safety and tolerability of PTI-125 at 100 mg twice-daily for 12 months. The study’s target enrollment is approximately 100 patients with mild-to-moderate Alzheimer’s disease, including patients from prior studies of PTI-125. Study sites may initially slow the pace of patient enrollment to minimize any risks of exposing elderly patients to infectious disease during office visits.
Future Clinical Plans
The basis for rational drug development is to ask and answer important questions around safety, tolerability and efficacy. The sequence of our smaller Phase 1 and Phase 2 studies are intended to generate preliminary conclusions around the safety and tolerability of PTI-125 for its intended indication to treat Alzheimer’s disease. Following the conclusion of these smaller studies, we may design and conduct a large-scale Phase 3 efficacy program of PTI-125 in Alzheimer’s disease in the future. Our strategy would be to tailor a Phase 3 efficacy program to the strengths and prior clinical observations of PTI-125.
In early 2018, FDA released a draft guidance document for Alzheimer’s drug development. While helpful, this draft guidance document does not provide a consensus opinion around the proper design of a Phase 3 efficacy program. In addition, the basic requirement to show a drug-placebo difference on a cognitive measure and a global or functional co-primary measurement appears to be unchanged. We believe our clinical efficacy program will need to correlate improvements in biomarkers with beneficial effects on cognition and or function in Alzheimer’s patients.
In order to take advantage of an evolving regulatory landscape, we intend to design and conduct a Phase 3 efficacy program in close collaboration with FDA and scientific, clinical and industry advisors. Any detailed consideration of our Phase 3 efficacy program is predicated on such collaboration. However, we believe current regulatory standards around the design and conduct of a Phase 3 study in Alzheimer’s disease may include the following baseline requirements:
|
· |
Study design: randomized, placebo-controlled, double-blind, multi-center study; |
|
· |
Objective: to assess the effects of PTI-125 on cognition, function and biomarkers; |
|
· |
Study size: 750 to 1,000 patients; |
|
· |
Patient population: mild to moderate Alzheimer’s disease, also including mild cognitively impaired; |
11
|
· |
Study duration: 18 to 24 months; |
|
· |
Primary endpoint: slower rate of decline over 18 months vs placebo on cognition and/or daily function; and |
|
· |
Co-primary endpoint: biomarkers of disease, hippocampal volume, others. |
Large-scale Phase 3 efficacy programs in Alzheimer’s disease are complex and very expensive, with greater challenges in planning, design, measurements methodology, implementation and analysis compared to smaller studies.
SavaDx is our investigational diagnostic to detect Alzheimer’s disease from a small sample of blood.
Our diagnostic effort, called SavaDx, is an early-stage program focused on detecting Alzheimer’s disease from a small sample of blood, possibly years before the overt appearance of clinical symptoms. The goal of SavaDx, an early-stage product candidate, is to make the detection of Alzheimer’s disease as simple as getting a blood test. We are developing SavaDx as a fast, accurate and quantitative blood-based biomarker/diagnostic to detect and monitor Alzheimer's disease. If successful, we believe SavaDx has potential to make obsolete more invasive approaches for diagnosing Alzheimer’s disease.
Over the past ten years, we discovered that altered FLNA is a hallmark feature of brain pathology in patients with Alzheimer’s disease. We believe SavaDx, which is a complex and unique detection system for altered filamin A (FLNA), can reveal early traces of the disease, potentially even before the overt appearance of disease symptoms, such as memory loss.
In September 2017, we announced a $1.8 million research grant award from the NIH for SavaDx. The NIH awarded us this research grant following a confidential, in-depth evaluation of SavaDx technology for scientific and technical merit. This technical milestone-based grant award enables us to work collaboratively with leaders in the field to collect clinical samples needed to develop our blood-based diagnostic for Alzheimer’s disease.
A diagnostic test usually measures one or more biomarkers, which are biological indicators of disease. A deep understanding of the biology of disease is required to identify and develop a diagnostic. A valid diagnostic has certain baseline characteristics to be functional and useful for clinical practice. It must detect disease in patients and, conversely, not detect disease in healthy subjects; and it is preferably quantitative, giving some indication of severity or stage of disease. Collectively, the ability to selectively detect disease indicators can be useful to provide diagnostic information (i.e., detect the disease) or prognostic information (i.e., predict the disease or its future course).
Currently, the most definitive method to diagnose Alzheimer’s disease is through autopsy after death, which is not particularly helpful. Methods to detect Alzheimer’s disease during its course can be expensive, invasive, subjective, risky or uncomfortable. Importantly, because of the expense and invasiveness of current tests, most people are not tested until they show obvious cognitive decline.
Current approaches for diagnosing Alzheimer’s disease include measurement of amyloid-β (specifically, Aβ42), total tau (T-tau) or phosphorylated tau (P-tau) levels in CSF; structural neuroimaging techniques, including magnetic resonance imaging (MRI) or computerized tomography (CT); positron-emission tomography (PET) imaging of brain amyloid (AmyVid®); and batteries of cognitive tests. Usually, a combination of more than one test is necessary to provide a working diagnosis. When such tests and techniques are used together, the totality of data can be sensitive and specific for the detection of Alzheimer’s disease. In practice, however, such tests and techniques are only used after overt symptoms of impaired memory.
We believe there is a profound need for a blood-based diagnostic test for Alzheimer’s disease. A quick, simple, inexpensive test may benefit the medical community in many ways. Advantages may include confirming the presence of Alzheimer’s disease earlier, when lifestyle changes and potential therapeutics may have the most impact, or conversely, to rule out Alzheimer’s disease at such early stages. Other potential benefits include discriminating Alzheimer’s disease from other causes of dementias; helping to identify stage of Alzheimer’s disease; selection and enrollment of appropriate patients into clinical studies of experimental product candidates; and better alignment of a patient’s specific diagnosis with a targeted therapeutic.
12
It is widely accepted that in Alzheimer’s disease, pathological changes in the brain occur at least 10-15 years before clinical symptoms appear. These “pre-symptomatic” changes include deposits of certain misfolded or impaired proteins in the brain. Our long-term goal with SavaDx is to identify people with Alzheimer’s disease, potentially long before clinical symptoms occur. Early detection may be critical for any intervention to cease - or at least slow down - brain damage before it is too late. Importantly, a non-invasive screen for latent Alzheimer’s disease prior to overt symptoms could be conducted as a general health screen, not just in patients at risk by family history or in patients already showing cognitive impairment. Once a disease-modifying treatment is found, early detection is likely to be critically important. Early detection and treatment may also be critical in identifying such a disease-modifying treatment, as many believe one reason for clinical study failures in Alzheimer’s disease is that treatment has routinely started too late in the course of disease to make any impact.
Moreover, with repeat measurements over time, SavaDx may provide a probability of cognitive decline or disease progression. Even if SavaDx does not provide a precise numerical cutoff value for Alzheimer’s disease, we believe it may be important to incorporate data from SavaDx into the overall diagnostic framework for neurodegeneration, and Alzheimer’s disease in particular. As with any diagnosis of disease, some people may embrace a way to detect Alzheimer’s disease long before clinical symptoms appear, while others may prefer not to know – at least until a treatment is found.
Diagnostic development program for SavaDx.
Diagnostic development differs from drug development in many important ways. As a result, diagnostic development requires substantial differences in planning, study design and study execution.
Some of the ways that diagnostic development differs from drug development include the following:
|
· |
We may need to choose among a wider range of regulatory pathways for approval of SavaDx, depending on factors such as intended use and user, test type and complexity and role in patient-care decisions; |
|
· |
Drug studies usually deal primarily with one office within FDA, but the regulatory pathway for SavaDx may require us to consider the policies of multiple federal or state regulatory agencies and offices; |
|
· |
Unlike drug programs, statistical analysis with SavaDx does not focus on efficacy and safety endpoints. Rather, study endpoints for SavaDx will focus on sensitivity (true positives), specificity (true negatives), positive predictive value (percentage of correct positive diagnoses of known positive cases) and negative predictive value (percentage of correct negative diagnoses of known negative cases); |
SavaDx is an investigational diagnostic product candidate that has not yet been reviewed by FDA. Clinical testing consists of collecting blood samples on a limited scale to validate SavaDx. Our ability to test such samples depends on multiple factors, many of which are beyond our control. For example, optimal sample collection depends on risk of sample degradation, storage requirements to preserve samples, cost of sample storage and actual vs. predicted time of assay validation.
Over the past three years, we have conducted four such rounds of early validation tests, with funding from NIH. In three blinded studies of test samples, SavaDx detected more than a 10-fold separation between Alzheimer’s patients and normal healthy control subjects (N=232 test samples). In these three proof-of-concept studies, SavaDx demonstrated nearly 100% accuracy and specificity. The three studies deployed a research grade antibody manufactured by an outside vendor.
A fourth blinded study of SavaDx failed to generate meaningful diagnostic data. We believe the fourth study deployed a faulty research antibody sourced from an outside vendor. Commercially available research antibodies can present certain technical flaws, such as improper validation, significant batch-to-batch variations or inconsistent storage, any of which can jeopardize results of studies and experiments. For these reasons, and in order to increase consistency of quality, reliability and availability, we are now developing and validating a proprietary, fit-for-purpose, antibody system for use with SavaDx. NIH is funding the development expenses for this project. Such technical activities are on-going. Assuming technical success with on-going efforts, we expect to run validation studies with SavaDx in the second half of 2020.
The legal system for intellectual property around diagnostic methods is highly complex and uncertain. In the U.S., patent courts have struggled to define a clear means of patent eligibility for modern age diagnostics. Generally, a simple process involving correlations between blood test results and patient health is not eligible for patent claims because such processes incorporate “laws of nature”. However, different outcomes from different courts, including Federal Circuit, district court and
13
Patent Trial and Appeal Board decisions, have continued to create a sometimes vague or conflicting legal framework for determining the eligibility of patent claims for diagnostic methods. As a result, we cannot be certain how SavaDx fits into the current U.S. legal framework for obtaining effective patent claims. Furthermore, claims for diagnostic methods can be complicated to enforce.
Expansion of our science to other indications.
It is well-known that protein misfolds occur in a wide variety of biological processes and diseases. We may leverage our scientific insights in neurodegeneration and advanced tools in biochemistry, bioinformatics and imaging to expand our science to other diseases. New indications and new drug development approaches may complement our initial focus on Alzheimer’s disease.
Pre-clinical programs are always visionary, sometimes innovative and often of high biomedical potential. However, by definition such programs are exploratory and risky. Moreover, most pre-clinical programs fail for scientific or other reasons, regardless of the amount of effort or resources that are brought to bear upon such programs. For these reasons, in general we do not intend to disclose our pre-clinical programs until such time as they become material to our pipeline of product candidates.
We own worldwide rights to our neurodegeneration program.
We own intellectual property, including patents, patent applications, technology, trade secrets and know-how in the U.S. and other countries. The protection of patents, designs, trademarks and other proprietary rights that we own or license is critical to our success and competitive position. We consider the overall protection of our patents and other intellectual property rights to be of material value and act to protect these rights from infringement.
We seek to protect our technology by, among other methods, filing and prosecuting U.S. and foreign patents and patent applications with respect to our technology and products and their uses. The focus of our patent strategy is to secure and maintain intellectual property rights to technology for our program in neurodegeneration.
PTI-125 and SavaDx were both discovered and designed in-house and were characterized by our academic collaborators during research activities that were conducted from approximately 2008 to date. We own exclusive, worldwide rights to these drug assets and related technologies, without royalty obligations to any third party. Our patent protection in this area currently runs through 2034, plus extensions, and includes six issued patents and related patent filings and applications.
Our Development Team
Our product development team is led by seasoned professionals with a proven track record of innovation in drug discovery and development, as well as substantial business expertise.
Our Founder and Chief Executive Officer, Remi Barbier, has over 25 years of biopharmaceutical industry experience and has led teams responsible for pioneering several pharmaceutical innovations, including abuse-deterrent drugs; the clinical development of multiple pain drugs; an innovative antibody program in cancer; and other programs in neuroscience and other therapeutics areas. Before founding Cassava Sciences (formerly known as Pain Therapeutics, Inc.), he held leadership roles and was founder or co-founder of four life science companies, three of which are now publicly traded. Our Chief Medical Officer, Nadav Friedmann PhD, MD, has eight prior FDA drug approvals and previously served as CEO of Daiichi Pharmaceuticals USA and Head of Johnson & Johnson’s Biotechnology Research Center. Lindsay Burns, PhD, SVP, Neuroscience, worked on the development of several product candidates in neuroscience and other therapeutics areas while at Neurex (acquired by Elan Pharmaceuticals) and Abgenix (acquired by Amgen). Michael Zamloot, SVP of Technology Operations, has four prior FDA drug approvals and has worked in drug operations and supply chain management at Boehringer Mannheim (acquired by Roche Diagnostics), Athena Neuroscience (acquired by Elan Pharmaceuticals) and Ciba-Geigy (acquired by Novartis). George (Ben) Thornton, PhD, SVP of Technology, has led research and development teams at Johnson & Johnson as well as translated basic science to the clinical setting at biotechnology start-ups such as GeneMedicine and Apovia.
Our management team is further supported by scientific advisors that share our commitment to advancing new treatments for Alzheimer’s disease. Leading experts in the field who advise us include:
14
|
· |
Jeff Cummings, MD, Director of Cleveland Clinic Lou Ruvo Center for Brain Health and Professor of Neurotherapeutics and Drug Development, Cleveland Clinic. |
|
· |
Hoau-Yan Wang, PhD, Tenured Medical Professor at CUNY Medical School; co-lead scientist on discovery & development of PTI-125 and SavaDx. |
|
· |
Steven E. Arnold, MD, Translational Neurology Head of the Interdisciplinary Brain Center, Massachusetts General Hospital, Harvard Medical School. |
|
· |
Barbara Sahakian, FBA, FMedSci, Professor of Clinical Neuropsychology at the Department of Psychiatry and Medical Research Council /Wellcome Trust Behavioral and Clinical Neuroscience Institute, University of Cambridge. |
|
· |
Trevor W. Robbins, CBE FRS FMedSci, Professor of Cognitive Neuroscience at the University of Cambridge and Past President of the British Neuroscience Association. |
Our Strategy
Our goal is to develop product candidates to diagnose and treat neurodegeneration, such as Alzheimer’s disease. Key elements of our business strategy to achieve this mission include:
|
· |
building a lean company that is narrowly focused on developing innovative product candidates for Alzheimer’s disease and other areas of neurodegeneration; |
|
· |
validating our unique scientific approach with competitive research grants and publishing our scientific data in peer-reviewed journals; |
|
· |
applying our development capabilities to advance our product candidates through clinical proof-of-concept studies and beyond; |
|
· |
using our expertise and experience to continue to focus on discovering new indications and product candidates, validated by experimental evidence and leading experts in the field; and |
|
· |
continuing to outsource preclinical studies, clinical studies and formulation development activities in order to allow more efficient deployment of our resources |
We also conduct basic research and development in collaboration with academic and other partners. Our research and development expenses were $1.6 million and $3.0 million for the year ended December 31, 2019 and 2018, respectively. These amounts are net of significant reimbursement received from NIH. See “Item 7 Management’s Discussion and Analysis of Financial Condition and Results of Operations” for additional details regarding our research and development activities.
Competition
The drug discovery and development industry is characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary products. We face potential competition from many different sources, including pharmaceutical and biotechnology companies, academic institutions and governmental agencies and public and private research institutions. Any product candidates that we successfully develop and commercialize, such as PTI-125 or SavaDx, may compete with existing therapies and new therapies that may become available in the future.
Historically, the drug industry has attempted to treat Alzheimer’s disease by developing drugs that block the synthesis of, or remove or dis-aggregate, beta amyloid and, more recently another protein in the brain called tau. Essentially, the prevailing doctrine says amyloid must be cleared out of the brain. This scientific approach – known as the amyloid hypothesis - has been repeatedly tested by our competitors in late stage clinical studies using a variety of antibody backbones, epitopes, target conformations, biomarkers and in various stages of disease. While this approach may yet work, to date the amyloid hypothesis has failed to generate unambiguous therapeutic benefit in patients with Alzheimer’s disease. More recent competitors in
15
Alzheimer’s research are focused on modulating proteins in the brain that have anti-inflammatory or other properties, an approach known as immunotherapy.
In contrast, our scientific approach seeks to simultaneously improve neurodegeneration and neuroinflammation. We believe improving multiple vital functions in the brain represents a new, different and crucial approach to address Alzheimer’s disease.
Regardless of scientific approach, improvements in cognition and function remains a key criterion for FDA approval of a new drug in Alzheimer’s disease, a hurdle which, to date, no drug candidate has met with clear and compelling clinical data.
Our competitors may have significantly greater financial resources, an established presence in the market, expertise in research and development, manufacturing, preclinical and clinical testing, obtaining regulatory approvals and reimbursement and marketing-approved products. These competitors also compete with us in recruiting and retaining qualified scientific and technical personnel, establishing clinical study sites and patient registration for clinical studies, as well as in acquiring or developing technologies complementary to, or necessary for, our programs. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies.
The key competitive factors affecting the success of PTI-125, and any other product candidates that we develop to address neurodegenerative disorders, if approved, are likely to be their efficacy, safety, convenience, price, the level of generic competition, patient and physician acceptance and the availability of reimbursement from government and other third-party payors. Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are more effective, have fewer or less severe side effects, are more convenient or are less expensive than any products that we may develop.
Our competitors also may obtain FDA approval for their products more rapidly than we may obtain approval for ours. For example, we are aware that Biogen, Inc., a large biopharmaceutical company, has announced their intention to seek regulatory approval in the U.S. for aducanumab in 2020. Aducanumab is a proprietary investigational drug candidate (human monoclonal antibody) that has been intensely studied for the treatment of Alzheimer’s disease. The safety and efficacy data with aducanumab are complex and subject to debate. However, aducanumab may become the first therapy since 2003 to receive FDA approval in patients with Alzheimer’s disease.
In recent years, we have observed ramped-up worldwide efforts aimed at developing blood-based techniques to detect and monitor Alzheimer’s disease. The key competitive factors affecting the success of SavaDx, and any other product candidates that we develop to diagnose neurodegeneration, if approved, are likely to be their measure of accuracy, such as specificity and sensitivity, as well as their convenience, patient acceptance, price and the availability of reimbursement from government and other third-party payors. Our competitors in the diagnostic area are pharmaceutical and biotechnology companies, academic institutions and governmental agencies and public and private research institutions. Despite increased research effort, the field has generally been hampered by lack of reproducibility and an unclear path on how to move academic discoveries into clinical utilization.
In addition to blood-based techniques to detect Alzheimer’s disease, competitors are examining the use of novel tracing agents and imaging techniques to map the course of neurodegeneration. In 2012 the FDA approved Amyvid® (Eli Lilly Pharmaceuticals), which is a radioactive diagnostic agent for brain imaging of amyloid plaque. Amyvid can rule out Alzheimer’s disease but does not confirm its presence. That is, a negative scan means little or no plaque is present; however, a positive scan does not necessarily indicate Alzheimer’s disease. In addition, Amyvid cannot be used to stage Alzheimer’s disease because some people take years to show cognitive decline after amyloid plaque develops, while other others rapidly develop advanced Alzheimer’s disease within months. Since its approval in 2012, Amyvid has had modest clinical utilization due to its high cost, lack of widespread reimbursement and need for specialized training.
Manufacturing
We do not own or lease any manufacturing facilities. We outsource formulation, manufacturing and related activities to third parties. For the foreseeable future, we will continue to rely on third parties to conduct certain quality control and assurance testing, shipping or storage of our product candidates.
16
We currently rely on contract development and manufacturing organizations (CDMOs) for the manufacture of all our materials for preclinical and clinical studies and expect to continue to do so, and for commercial supply of any product candidates that we may develop. We currently have established relationships with several CDMOs for the manufacturing of our product candidates.
Our suppliers must comply with current good manufacturing practices (cGMP) enforced by the FDA and other government agencies. Our suppliers are subject to unannounced inspection by regulators, including pre-approval inspections by the FDA, to ensure they are in strict compliance with government regulations and standards. Our suppliers may be forced to stop producing, storing, shipping or testing our drug products if they fall out of compliance with government regulations and standards.
We have no control over our suppliers’ compliance, or lack thereof, with the multitude of regulations and standards that affect our drug products. We cannot control decisions by our suppliers that affect their ability or willingness to continue to supply us on acceptable terms, or at all.
Strategic Shift Away from Analgesic Drug Development
Historically, our focus had been on analgesic drug development. During that time, we conceived, formulated, developed, tested and patented various analgesic drug candidates. In late 2018, we announced a strategic shift away from analgesic drug development. On March 20, 2019, we gave a prior collaborator, Durect Corporation, written notice of termination of a licensing agreement for analgesic drugs. This action effectively ended our involvement with analgesic drug development. In March 2019, we announced that going forward we would focus exclusively on our on-going program in neurodegeneration. On or around that time, we also announced our new company name, logo, NASDAQ ticker symbol and website.
Government Regulation
Government authorities in the U.S. at the federal, state and local level and in other countries regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing and export and import of drug and diagnostic products. Generally, before a new drug or diagnostic can be marketed, considerable data demonstrating its quality, safety and efficacy and/or specificity must be obtained, organized into a format specific for each regulatory authority, submitted for review and approved by the regulatory authority.
U.S. Drug Development
In the U.S., the FDA regulates drugs under the Food, Drug, and Cosmetic Act (FDCA). Both drugs and diagnostics also are subject to other federal, state and local statutes and regulations. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations requires the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or post-market may subject an applicant to administrative or judicial sanctions. These sanctions could include, among other actions, the FDA’s refusal to approve pending applications, withdrawal of an approval, a clinical hold, untitled or warning letters, product recalls or market withdrawals, product seizures, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement and civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on us.
Product candidates must be approved by the FDA before they may be commercialized in the U.S. The drug approval process generally involves the following:
|
· |
Completion of extensive preclinical studies in accordance with applicable regulations, including studies conducted in accordance with good laboratory practice; |
|
· |
Submission to the FDA of an IND, which must become effective before human clinical studies may begin; |
|
· |
Approval by an independent institutional review board (IRB) or ethics committee before each study may be initiated; |
17
|
· |
Performance of adequate and well-controlled human clinical studies in accordance with applicable IND regulations, code of good clinical practice (cGCP), requirements and other clinical trial-related regulations to establish the safety and efficacy of the investigational product for each proposed indication; |
|
· |
Submission to the FDA of an NDA; |
|
· |
A determination by the FDA within 60 days of its receipt of an NDA to accept the filing for review; |
|
· |
Satisfactory completion of an FDA pre-approval inspection of the manufacturing facility or facilities where the drug will be produced to assess compliance with cGMP, requirements to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; |
|
· |
Potential FDA audit of the preclinical study and/or clinical study sites that generated the data in support of the NDA; |
|
· |
FDA review and approval of the NDA, including consideration of the views of any FDA advisory committee, prior to any commercial marketing or sale of the drug in the U.S.; and |
|
· |
Compliance with any post-approval requirements, including the potential requirement to conduct post-approval studies. |
The data required to support an NDA are generated in two distinct developmental stages: preclinical and clinical. The preclinical and clinical testing and approval process requires substantial time, effort and financial resources, and we cannot be certain that any approvals for any future product candidates will be granted on a timely basis, or at all.
Preclinical Studies and IND
The preclinical developmental stage generally involves laboratory evaluations of drug chemistry, formulation and stability, as well as studies to evaluate toxicity in animals, which support subsequent clinical testing. As sponsor, we must submit the results of the preclinical studies, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, to the FDA as part of the IND. An IND is a request for authorization from the FDA to administer an investigational product to humans and must become effective before human clinical studies may begin.
Preclinical studies include laboratory evaluation of product chemistry and formulation, as well as in vitro and animal studies to assess the potential for adverse events and in some cases to establish a rationale for therapeutic use. The conduct of preclinical studies is subject to federal regulations and requirements, including cGCP regulations for safety/toxicology studies. An IND sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and plans for clinical studies, among other things, to the FDA as part of an IND. Some long-term preclinical testing, such as long-term toxicity tests, animal tests of reproductive adverse events and carcinogenicity, may continue after the IND is submitted. An IND automatically becomes effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions about any aspect of the program. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical study can begin.
Clinical Studies
The clinical stage of development involves the administration of the investigational product to healthy volunteers or patients under the supervision of qualified investigators, generally physicians not employed by or under the study sponsor’s control, in accordance with cGCP requirements, which include the requirement that all research subjects provide their informed consent for their participation in any clinical trial. Clinical studies are conducted under protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection and exclusion criteria and the parameters to be used to monitor subject safety and assess efficacy. Each protocol, and any subsequent amendments to the protocol, must be submitted to the FDA as part of the IND. Furthermore, each clinical study must be reviewed and approved by an IRB for each institution at which the clinical study will be conducted to ensure that the risks to individuals participating in the clinical studies are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the informed consent form that must be provided to each clinical study subject or his or her legal representative and must monitor the clinical study until
18
completed. There also are requirements governing the reporting of ongoing clinical studies and completed clinical study results to public registries.
A sponsor who wishes to conduct a clinical study outside of the U.S. may, but need not, obtain FDA authorization to conduct the clinical study under an IND. If a foreign clinical study is not conducted under an IND, the sponsor may submit data from the clinical study to the FDA in support of an NDA. The FDA may accept a well-designed and well-conducted foreign clinical study not conducted under an IND if the study was conducted in accordance with cGCP requirements and the FDA is able to validate the data through an onsite inspection if deemed necessary. In 2019 we did not conduct any clinical studies outside of the U.S. but we may do so in the future.
Clinical studies in the U.S. generally are conducted in three sequential phases, known as Phase 1, Phase 2 and Phase 3, and may overlap.
|
· |
Phase 1 clinical studies generally involve a small number of healthy volunteers or disease-affected patients who are initially exposed to a single dose and then multiple doses of the product candidate. The primary purpose of these clinical studies is to assess the metabolism, pharmacologic action, tolerability and safety of a drug candidate. |
|
· |
Phase 2 clinical studies involve studies in disease-affected patients to determine the proper dose required to produce the desired benefits. At the same time, safety and further pharmacokinetic and pharmacodynamic information is collected, possible adverse effects and safety risks are identified, and a preliminary evaluation of efficacy may be observed. |
|
· |
Phase 3 clinical studies generally involve many patients at multiple sites and are designed to provide the data necessary to demonstrate the effectiveness of the product for its intended use, its safety in use and to establish the overall benefit/risk relationship of the product and provide an adequate basis for product approval. These studies may include comparisons with placebo and/or other comparator treatments. The duration of treatment is often extended to mimic the actual use of a product during marketing. |
Post-approval studies, sometimes referred to as Phase 4 clinical studies, may be conducted after initial marketing approval. These studies are used to gain additional experience from the treatment of patients in the intended therapeutic indication. In certain instances, the FDA may mandate the performance of Phase 4 clinical studies as a condition of approval of an NDA.
Progress reports detailing the results of the clinical studies, among other information, must be submitted at least annually to the FDA. Written safety reports and the investigators for serious and unexpected adverse events, or any other findings suggesting a significant risk to humans exposed to the drug must be submitted to the FDA.
Phase 1, Phase 2, and Phase 3 clinical studies may not be completed successfully within any specified period, if at all. The FDA or the sponsor may suspend or terminate a clinical study at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical study at its institution if the clinical study is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients. Additionally, some clinical studies are overseen by an independent group of qualified experts organized by the clinical study sponsor, known as a data safety monitoring board or committee. This group provides authorization for whether a study may move forward at designated check-points based on access to certain data from the trial. Concurrent with clinical studies, companies usually complete additional animal studies and must develop additional information about the chemistry and physical characteristics of the drug as well as finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product and, among other things, companies must develop methods for testing the identity, strength, quality and purity of the final product. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that our product candidates do not undergo unacceptable deterioration over their shelf life.
19
NDA Review Process
Following completion of the clinical studies, data is analyzed to assess whether the investigational product is safe and effective for the proposed indicated use or uses. The results of preclinical studies and clinical studies are then submitted to the FDA as part of an NDA, along with proposed labeling, chemistry and manufacturing information to ensure product quality and other relevant data. In short, the NDA is a request for approval to market a drug for one or more specified indication and must contain proof of safety and efficacy for a drug’s purity and potency. The application may include both negative and ambiguous results of preclinical studies and clinical studies, as well as positive findings. Data may come from company-sponsored clinical studies intended to test the safety and efficacy of a product’s use or from several alternative sources, including studies initiated by investigators. To support marketing approval, the data submitted must be sufficient in quality and quantity to establish the safety and efficacy of the investigational product to the satisfaction of FDA. FDA approval of an NDA must be obtained before a drug may be marketed in the U.S.
Under the Prescription Drug User Fee Act (PDUFA), as amended, each NDA must be accompanied by a user fee. FDA adjusts the PDUFA user fees on an annual basis. According to the FDA’s fiscal year 2020 fee schedule, effective through September 30, 2020, the user fee for an application requiring clinical data, such as an NDA, is approximately $2.9 million. Fee waivers or reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a small business. Additionally, no user fees are assessed on NDAs for products designated as orphan drugs, unless the product also includes a non-orphan indication.
The FDA reviews all submitted NDAs before it accepts them for filing and may request additional information rather than accept the NDA for filing. The FDA must decide on accepting an NDA for filing within 60 days of receipt. Once the submission is accepted for filing, the FDA begins an in-depth review of the NDA. Under the goals and policies agreed to by the FDA under PDUFA, the FDA has 10 months, from the filing date, in which to complete its initial review of a new molecular-entity NDA and respond to the applicant, and six months from the filing date of a new molecular-entity NDA designated for priority review. The FDA does not always meet its PDUFA goal dates for standard and priority NDAs, and the review process is often extended by FDA requests for additional information or clarification.
Before approving an NDA, the FDA may conduct a pre-approval inspection of the manufacturing facilities for the new product to determine whether they comply with cGMP requirements. The FDA will not approve the product unless it determines that the manufacturing processes and facilities fully comply with cGMP requirements and are adequate to assure consistent production of the product within required specifications. The FDA also may audit data from clinical studies to ensure compliance with cGCP requirements. Additionally, the FDA may refer applications for novel product candidates which present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions, if any. The FDA is not bound by recommendations of an advisory committee, but it considers such recommendations when making decisions on approval. The FDA likely will reanalyze the clinical study data, which could result in extensive discussions between the FDA and the applicant during the review process. After the FDA evaluates an NDA, it will issue either an approval letter or a Complete Response Letter (CRL). An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications. A CRL indicates that the FDA’s review of the application is complete and the application cannot be approved in its present form. A CRL usually describes the specific deficiencies in the NDA identified by the FDA. The CRL may require additional clinical data, additional pivotal Phase 3 clinical trial(s) and/or other significant and time-consuming requirements related to clinical studies, preclinical studies or manufacturing. If a CRL is issued, the applicant may either resubmit the NDA, addressing all the deficiencies identified in the CRL, or withdraw the application. Even if such data and information are submitted, the FDA may decide that the NDA does not satisfy the criteria for approval. Data obtained from clinical studies are not always conclusive and the FDA may interpret data differently than we interpret the same data.
Advertising and Promotion
The FDA and other federal regulatory agencies closely regulate the marketing and promotion of drugs through, among other things, standards and regulations for direct‑to‑consumer advertising, communications regarding unapproved uses, industry‑sponsored scientific and educational activities, and promotional activities involving the Internet. None of our product candidates can be commercially promoted before receiving FDA approval. After approval, product promotion can include only those claims relating to safety and effectiveness that are consistent with the labeling approved by the FDA. Healthcare providers are permitted to prescribe drugs for “off‑label” uses — that is, uses not approved by the FDA and therefore not
20
described in the drug’s labeling — because the FDA does not regulate the practice of medicine. However, FDA regulations impose stringent restrictions on manufacturers’ communications regarding off‑label uses. Failure to comply with applicable FDA requirements and restrictions in this area may subject us to adverse publicity and enforcement action by the FDA, the U.S. Department of Justice, or the Office of the Inspector General of Health and Human Services, as well as state authorities. This could subject us to a range of penalties that could have a significant commercial impact, including civil and criminal fines and agreements that materially restrict the manner in which we promote or distribute our product candidates.
Post‑Approval Requirements
After a product candidate receives regulatory approval, it is often subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to drug listing and registration, recordkeeping, periodic reporting, product sampling and distribution, adverse event reporting and advertising, marketing and promotion restrictions.
Adverse event reporting and submission of periodic reports is required following FDA approval of an NDA. The FDA also may require post‑market testing, known as Phase 4 testing, or the FDA may place conditions on an approval that could restrict the distribution or use of the product. In addition, quality control, drug manufacture, packaging, and labeling procedures must continue to conform to cGMP after approval. Drug manufacturers and certain of their subcontractors are required to register their establishments with the FDA and certain state agencies. Registration may result in periodic announced or unannounced inspections by the FDA or these state agencies, during which the agency inspects manufacturing facilities to assess compliance with cGMP. Accordingly, manufacturers must continue to expend time, money, and effort in the areas of production and quality control to maintain compliance with cGMP. Regulatory authorities may withdraw product approvals or request product recalls if a company fails to comply with regulatory standards, if it encounters problems following initial marketing, or if previously unrecognized problems are subsequently discovered. In addition, other regulatory actions may be taken, including, among other things, warning letters, the seizure of products, injunctions, consent decrees placing significant restrictions on or suspending manufacturing operations, refusal to approve pending applications or supplements to approved applications, civil penalties, and criminal prosecution.
The FDA may require post‑approval clinical studies to help assure continued safety or effectiveness of the approved drug. The FDA may also require a labeling change if it becomes aware of new safety information that it believes should be included in the labeling of a drug.
The Hatch‑Waxman Amendments
Orange Book Listing
In seeking approval for our product candidates through an NDA, we will be required to list with the FDA each patent whose claims cover the drug product. Upon receiving regulatory approval, each of the patents listed in the application for this drug is then published in the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the “Orange Book”. Drugs listed in the Orange Book can, in turn, be cited by potential generic competitors in support of approval of an abbreviated NDA, or ANDA. An ANDA provides for marketing of a drug product that has the same active ingredient in the same strengths and dosage form as the listed drug and has been shown through bioequivalence testing to be therapeutically equivalent to the listed drug. Other than the requirement for bioequivalence testing, ANDA applicants are not required to conduct, or submit results of, preclinical or clinical tests to prove the safety or efficacy of their drug product. Drugs approved in this way are commonly referred to as “generic equivalents” to the listed drug, and can often be substituted by pharmacists under prescriptions written for the original listed drug.
The ANDA applicant is required to make certain certifications to the FDA concerning any patents listed for the approved product in the FDA’s Orange Book. Specifically, the applicant must certify that: (i) the required patent information has not been filed; (ii) the listed patent has expired; (iii) the listed patent has not expired but will expire on a particular date and approval is sought after patent expiration; or (iv) the listed patent is invalid or will not be infringed by the new product. The ANDA applicant may also elect to submit a section viii statement certifying that its proposed ANDA label does not contain (or carves out) any language regarding the patented method‑of‑use rather than make certifications concerning a listed method‑of‑use patent. If the applicant does not challenge the listed patents, the ANDA application will not be approved until all the listed patents claiming the referenced product have expired.
21
A certification that the new product will not infringe the already approved product’s listed patents, or that such patents are invalid, is called a Paragraph IV certification. If the ANDA applicant has provided a Paragraph IV certification to the FDA, the applicant must also send notice of the Paragraph IV certification to the NDA and patent holders once the ANDA has been accepted for filing by the FDA. The NDA and patent holders may then initiate a patent infringement lawsuit in response to the notice of the Paragraph IV certification. The filing of a patent infringement lawsuit within 45 days of the receipt of a Paragraph IV certification automatically prevents the FDA from approving the ANDA until the earlier of 30 months, expiration of the patent, settlement of the lawsuit, or a decision in the infringement case that is favorable to the ANDA applicant. The ANDA application also will not be approved until any applicable non‑patent exclusivity listed in the Orange Book for the referenced product has expired.
Exclusivity
Upon NDA approval of a new chemical entity, or NCE, which is a drug that contains no active moiety that has been approved by the FDA in any other NDA, that drug receives five years of marketing exclusivity during which the FDA cannot receive any ANDA seeking approval of a generic version of that drug or any Section 505(b)(2) NDA that relies on the FDA’s findings regarding that drug. A drug may obtain a three‑year period of exclusivity for a change to the drug, such as the addition of a new indication to the labeling or a new formulation, during which the FDA cannot approve an ANDA or any Section 505(b)(2) NDA, if the supplement includes reports of new clinical studies (other than bioavailability clinical studies) essential to the approval of the supplement.
An ANDA may be submitted one year before NCE exclusivity expires if a Paragraph IV certification is filed. If there is no listed patent in the Orange Book, there may not be a Paragraph IV certification, and, thus, no ANDA may be filed before the expiration of the exclusivity period.
Disclosure of Clinical Study Information
Sponsors of clinical studies of FDA‑regulated products, including drugs, are required to register and disclose certain clinical study information. Information related to the product, patient population, phase of investigation, clinical study sites and investigators, and other aspects of the clinical study is then made public as part of the registration. Sponsors are also obligated to post certain information regarding the results of their clinical studies after completion. Disclosure of the results of these studies can be delayed until the new product or new indication being studied has been approved. Competitors may use this publicly available information to gain knowledge regarding the progress of development programs.
Other Regulatory Requirements
We may be subject to federal, state and local environmental laws and regulations, including the Environmental Protection Act and the Clean Air Act. Although we believe that our safety procedures for handling and disposing of controlled materials comply with the standards prescribed by state and federal regulations, accidental contamination or injury from these materials may occur. In the event of such an occurrence, we could be held liable for any damages that result and any such liability could exceed our resources.
We may also be subject to regulations under other federal, state, and local laws, including the Occupational Safety and Health Act, national restrictions on technology transfer, and import, export, and customs regulations. It is possible that any portion of the regulatory framework under which we operate may change and that such change could have a negative impact on our current and anticipated operations. Failure to comply with these requirements could result, among other things, in suspension of regulatory approval, recalls, injunctions or civil or criminal sanctions.
Third‑Party Payor Coverage and Reimbursement
The commercial success of our product candidates, if approved, will depend, in part, upon the availability of coverage and adequate reimbursement from third‑party payors at the federal, state and private levels. Third‑party payors include governmental programs such as Medicare or Medicaid, private insurance plans and managed care plans. These third‑party payors may deny coverage or reimbursement for our product candidates in whole or in part if they determine that our product candidates are not medically appropriate or necessary. Also, third-party payors attempt to control costs by limiting coverage through the use of formularies and other cost‑containment mechanisms and the amount of reimbursement for particular procedures or drug treatments.
22
Some third‑party payors also require pre‑approval of coverage for new or innovative devices or drug therapies before they will reimburse healthcare providers who use such therapies. While we cannot predict whether any proposed cost‑containment measures will be adopted or otherwise implemented in the future, these requirements or any announcement or adoption of such proposals could have a material adverse effect on our ability to obtain adequate prices for our approved product candidates to operate profitably.
Employees
As of December 31, 2019, we had 9 full-time employees. None of our employees are represented by a labor union or covered under a collective bargaining agreement. We also engage numerous consultants to perform services on retainer, per diem or an hourly basis.
Corporate Information
We were incorporated as a Delaware corporation in May 1998 under the name Pain Therapeutics, Inc. On March 26, 2019, we changed our company name to Cassava Sciences, Inc. Our principal offices are located at 7801 N. Capital of Texas Highway, Suite 260, Austin, TX, 78731. Our telephone number is 512-501-2444. Our website address is www.CassavaSciences.com.
We use Cassava Sciences, the Cassava Sciences logo, artwork and other marks as trademarks in the United States and other countries. Solely for convenience, trademarks and trade names referred to in this Annual Report, including logos, artwork, and other visual displays, may appear without the ® or TM symbols, but such references are not intended to indicate in any way that we will not assert, to the fullest extent under applicable law, our rights, or the rights of the applicable licensor to these trademarks and trade names. We do not intend our use or display of other entities’ trade names, trademarks, or service marks to imply a relationship with, or endorsement or sponsorship of us by, any other entity.
We file electronically with the Securities and Exchange Commission, or SEC, our annual reports on Form 10-K, quarterly reports on Form 10-Q and current reports on Form 8-K pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934. The SEC maintains an Internet site that contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC. The address of the site is http://www.sec.gov.
You may obtain a free copy of our annual reports on Form 10-K, quarterly reports on Form 10-Q and current reports on Form 8-K and amendments to those reports on the day of filing with the SEC on our website at http://www.cassavasciences.com, by contacting our corporate offices by calling 512-501-2450 or by sending an e-mail message to IR@cassavasciences.com.
23
RISK FACTORS
Investing in our common stock involves a high degree of risk. You should carefully consider the risks described below, as well as other information contained in this Annual Report on Form 10-K, including our financial statements and the related notes and the section titled “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” before deciding whether to invest in our common stock. The occurrence of any of the events or developments described below could harm our business, financial condition, results of operations, and growth prospects. In such an event, the market price of our common stock could decline, and you may lose all or part of your investment.
Additional risks and uncertainties not presently known to us or that we currently deem immaterial also may impair our business operations and the market price of our common stock.
Risks Related to Our Business, Financial Condition, and Capital Requirements
We may need to cease our operations if we are unable to retain key personnel.
We are engaged in developing early-stage technologies and will continue to do so for the foreseeable future. Unlike larger organizations, we rely on a very small number of highly skilled, and highly sought after, employees to continue the advancement of our development stage technologies. The knowledge and skills contributed by our key employees may be irreplaceable and the loss of a key employee may cause substantial negative financial, operational and scientific consequences for our business. As an example, our research grant awards from NIH depend in part on the continued participation of certain key employees, known as a Principal Investigator. The loss of a Principal Investigator may result in the loss of one or more research grant awards from NIH, which would have significant adverse effects on our ability to continue to conduct, conclude or fund our research programs in Alzheimer’s disease. Likewise, the intellectual property that is intended to protect our development stage technologies is still evolving and its evolution remains highly dependent on a small number of employees with specific expertise. The loss of a key employee may jeopardize our existing or pending intellectual property or may prevent us from accessing the technical information and knowledge necessary to extend our portfolio of intellectual property. Furthermore, we believe the adverse effects that may result from losing a key employee’s participation cannot be compensated with any specific insurance policies, such as “key person” or “business life” insurance. If we are not successful in retaining key employees, our business and financial condition will suffer, and we may need to cease our operations.
We will need substantial funding to advance PTI-125 into a Phase 3 clinical efficacy program in Alzheimer’s disease.
We believe our future success is highly dependent upon our ability to successfully advance PTI-125 in a Phase 3 clinical efficacy program in patients with Alzheimer’s disease. Phase 3 programs in Alzheimer’s are complex and costly, sometimes exceeding $100 million in the aggregate over a period of years. We currently do not have sufficient cash-on-hand to fund a Phase 3 program of PTI-125 in Alzheimer’s disease, nor can we readily anticipate having access to such funding in the future. If access to sufficient funds does become available to us in the future, the terms and conditions may be so onerous that current stockholders will suffer substantial dilution or loss of investment. If we cannot access sufficient funds, we will not be able to advance PTI-125 in a Phase 3 clinical efficacy program and our business, results of operations and financial condition will suffer.
We may not be successful in developing our product candidates in neurodegeneration.
Our product candidates in neurodegeneration are still in the early stages of development and will take several more years to develop and must undergo extensive clinical and scientific validations. Even if we are successful in developing any of our product candidates through clinical and scientific validation, we may not be able to develop a drug or a diagnostic that:
|
· |
meets applicable regulatory standards, in a timely manner or at all; |
|
· |
successfully competes with other technologies and tests; |
|
· |
avoids infringing the proprietary rights of others; |
24
|
· |
are adequately reimbursed by third-party payors; |
|
· |
can be performed at commercial levels or at reasonable cost; or |
|
· |
can be successfully marketed. |
To the extent we are not successful in developing our new product candidates in neurodegeneration, our results of operations and business will be materially adversely affected.
We have a limited operating history in our business targeting Alzheimer’s disease and no history of product approvals for commercial sale, which may make it difficult to evaluate our current business and predict our future success and viability.
We are an early clinical-stage biopharmaceutical company with a limited operating history in our business targeting Alzheimer’s disease. Since we commenced operations in May 1998, we have had no product candidates approved for commercial sale and have not generated any revenue from product sales. Drug development is a highly uncertain undertaking and involves a substantial degree of risk. To date, we have not initiated or completed a pivotal clinical study involving Alzheimer’s disease, obtained marketing approval for any product candidates, or conducted sales and marketing activities necessary for successful product commercialization. Our long operating history as a company without product revenue makes any assessment of our future success and viability subject to significant uncertainty.
We will continue to encounter risks and difficulties frequently experienced by early-stage biopharmaceutical companies in rapidly evolving fields. We have not yet demonstrated an ability to successfully overcome such risks and difficulties. If we do not successfully address these risks and difficulties, our business, results of operations and financial condition will suffer materially.
In addition, from inception through 2018 our principal activity was analgesic drug development. In late 2018, we announced a strategic shift away from analgesic drug development to focus on neurodegeneration. Going forward, and for the foreseeable future, we anticipate that our product candidates for neurodegeneration will be the principal focus of our business operations and such efforts will consume the vast majority of our resources. We may not be successful in transitioning to the development of product candidates for neurodegeneration, such as Alzheimer’s disease.
We have incurred significant net losses in each period since our inception and anticipate that we will continue to incur net losses for the foreseeable future.
We have incurred net losses in each reporting period since our inception, including a net loss of $4.6 million for the year ended December 31, 2019. As of December 31, 2019, we had an accumulated deficit of $168.6 million.
We have invested significant financial resources in research and development activities for product candidates. We do not expect to generate revenue from product sales for several years, if at all. The amount of our future net losses will depend, in part, on the level of our future expenditures and revenue. Moreover, our net losses may fluctuate significantly from quarter to quarter and year to year, such that a period-to-period comparison of our results of operations may not be a good indicator of our future performance.
We expect to continue to incur significant expenses and higher operating losses for the foreseeable future. We anticipate that our expenses will increase substantially as we:
|
· |
continue our research and discovery activities; |
|
· |
advance our current and any future product candidates through preclinical and clinical development; |
|
· |
initiate and conduct additional preclinical, clinical, or other studies for our product candidates; |
|
· |
work with our CDMO’s to scale up the manufacturing processes for our product candidates; |
|
· |
seek regulatory approvals and marketing authorizations for our product candidates; |
25
|
· |
obtain, maintain, protect, defend and enforce our intellectual property portfolio; |
|
· |
attract, hire, and retain qualified personnel; |
|
· |
provide additional internal infrastructure to support our continued research and development operations and any planned commercialization efforts in the future; |
|
· |
experience any delays or encounter other issues related to our operations; and |
|
· |
meet the requirements and demands of being a public company. |
Our prior losses and expected future losses have had and will continue to have an adverse effect on our stockholders’ equity and working capital. In any quarter, our operating results could be below the expectations of securities analysts or investors, which could cause our stock price to decline.
We have no product revenues and may never achieve revenues or profitability based on product revenues.
We have no products approved for commercial sale. To obtain revenues from the sales of our product candidates that are significant or large enough to achieve profitability, we must succeed, either alone or with third parties, in developing, obtaining regulatory approval for, manufacturing, and marketing product candidates with significant commercial value. This is a significant endeavor that few early-stage biopharmaceutical companies can successfully achieve. Our ability to generate revenue and achieve profitability depends on many factors, including:
|
· |
completing research and preclinical and clinical development of our product candidates; |
|
· |
obtaining regulatory approvals and marketing authorizations for product candidates for which we successfully complete clinical development; |
|
· |
developing a sustainable and scalable manufacturing process for our product candidates, as well as establishing and maintaining commercially viable supply relationships with third parties that can provide adequate products and services to support clinical activities and commercial demand for our product candidates; |
|
· |
identifying, assessing, acquiring, and/or developing new product candidates; |
|
· |
negotiating favorable terms in any collaboration, licensing, or other arrangements into which we may enter; |
|
· |
addressing any competing technological and market developments; |
|
· |
maintaining, protecting, expanding, and enforcing our portfolio of intellectual property rights, including patents, trade secrets, and know-how; and |
|
· |
attracting, hiring, and retaining qualified personnel. |
Because of the numerous risks and uncertainties associated with drug development, we are unable to predict the timing or amount of our expenses, or when we will be able to generate any meaningful revenue or achieve or maintain profitability, if ever. In addition, our expenses could increase beyond our current expectations if we are required by the FDA or foreign regulatory agencies to perform studies in addition to those that we currently anticipate, or if there are any delays in any of our clinical studies or the development of any of our product candidates.
We will require additional capital to fund our operations and to complete the development of our product candidates. A failure to obtain this necessary capital on acceptable terms, or at all, could force us to delay, limit, reduce, or terminate our commercialization efforts, product development, or other operations.
Our operations have required substantial amounts of cash since inception, and we expect our expenses to increase significantly in the foreseeable future. To date, we have financed our operations primarily through the sale of equity securities,
26
research grants and payments received from prior third-party collaborations. Developing our product candidates and conducting clinical studies for the treatment of neurodegenerative diseases, including Alzheimer’s disease, will require substantial amounts of capital. We will also require a significant amount of capital to commercialize any approved products.
As of December 31, 2019, we had cash and cash equivalents of $23.1 million. Based on our current operating plan, we believe that our existing cash and cash equivalents will be sufficient to fund our projected operations for at least the next 12 months. Our estimate as to how long we expect our existing cash and cash equivalents to be available to fund our operations is based on assumptions that may prove inaccurate, and we could use our available capital resources sooner than we currently expect. In addition, changing circumstances may cause us to increase our spending significantly faster than we currently anticipate, and we may need to spend more money than currently expected because of circumstances beyond our control. We may need to raise additional funds sooner than we anticipate if we choose to expand more rapidly than we presently anticipate.
We will require additional capital for the further development of our product candidates. Additional capital may not be available when we need it, or on terms acceptable to us or at all. We have no committed source of additional capital. If adequate capital is not available to us on a timely basis, we may be required to significantly delay, limit, reduce or terminate our research and development programs or the commercialization of product candidates, if approved, or be unable to continue or expand our operations, or otherwise capitalize on our business opportunities, as desired, which could materially affect our business, financial condition, results of operations, and growth prospects and cause the price of our common stock to decline.
To the extent that we raise additional capital through the sale of equity or convertible debt securities, the ownership interest of our stockholders will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect your rights as a common stockholder. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures, or declaring dividends. If we raise additional funds through collaborations, strategic alliances, or licensing arrangements with pharmaceutical partners, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs, or product candidates, or grant licenses on terms that may not be favorable to us.
Global credit and financial market conditions could negatively impact the value of our portfolio of cash equivalents, short-term investments or long-term investments and our ability to meet our financing objectives.
Our cash and cash equivalents are generally maintained in highly liquid investments with original maturities of 90 days or less at the time of purchase. Our short-term investments, if any, consist primarily of readily marketable debt securities with original maturities of greater than 90 days from the date of purchase but remaining maturities of less than one year from the balance sheet date. Our long-term investments, if any, consist primarily of readily marketable debt securities with maturities in one year or beyond from the balance sheet date. While, as of the date of this filing, we are not aware of any downgrades, material losses, or other significant deterioration in the fair value of our cash equivalents, short-term investments or long-term investments since December 31, 2019, no assurance can be given that deterioration in conditions of the global credit and financial markets would not negatively impact our current portfolio of cash equivalents, short-term investments or long-term investments or our ability to meet our financing objectives.
New accounting pronouncements and legislative actions may significantly impact our future financial position or results of operations.
Future changes in financial accounting standards may cause adverse, unexpected fluctuations in the timing of the recognition of revenues or expenses and may affect our financial position or results of operations. New pronouncements and varying interpretations of existing pronouncements have occurred with frequency, and may occur again in the future, which may require us to make changes in our accounting policies in the future. Compliance with changing regulation of corporate governance and public disclosure may result in additional expenses. Changing laws, regulations and standards relating to corporate governance and public disclosure, including the Sarbanes-Oxley Act of 2002 (SOX), new SEC regulations, PCAOB pronouncements and Nasdaq rules, are creating uncertainty for companies such as ours and insurance, accounting and auditing costs are high as a result of this uncertainty and other factors. We are committed to maintaining high standards of corporate governance and public disclosure. As a result, we intend to invest all reasonably necessary resources to comply with evolving standards, and this investment may result in increased general and administrative expenses and a diversion of management time and attention to compliance activities.
27
Risks Related to the Discovery, Development, and Commercialization of Our Product Candidates
Research and development of biopharmaceutical products is very risky. Our business is heavily dependent on the successful development of our product candidates, which are all in the early stages of development. We cannot give any assurance that any of our product candidates will receive regulatory approval, which is necessary before they can be commercialized.
We are at the early stages of development of our product candidates. To date, we have invested substantial effort and financial resources to identify, procure intellectual property for, and develop our programs in neurodegeneration, including conducting preclinical studies and early-stage clinical studies for our product candidates, PTI-125 and SavaDx, and providing general and administrative support for these operations. Our future success is dependent on our ability to successfully develop, obtain regulatory approval for, and then successfully commercialize our product candidates, and we may fail to do so for many reasons, including the following:
|
· |
our product candidates may not successfully complete preclinical studies or clinical studies; |
|
· |
a product candidate may, on further study, be shown to have harmful side effects or other characteristics that indicate it is unlikely to be effective or otherwise does not meet applicable regulatory criteria; |
|
· |
our competitors may develop products that render our product candidates obsolete or less attractive; |
|
· |
the product candidates that we develop may not be sufficiently covered by intellectual property; |
|
· |
the product candidates that we develop may be challenged by third parties’ patents or other intellectual property or exclusive rights; |
|
· |
the market for our product candidates may change so that the continued development of a product candidate is no longer reasonable or commercially attractive; |
|
· |
our product candidate may not be capable of being produced in commercial quantities at an acceptable cost, or at all; |
|
· |
if a product candidate obtains regulatory approval, we may be unable to establish sales and marketing capabilities, or successfully market such approved product candidate, to gain market acceptance; and |
|
· |
a product candidate may not be accepted as safe, effective or useful by patients, the medical community or third-party payors, if applicable. |
If any of these events occur, we may be forced to abandon our development efforts for a program or programs, which would have a material adverse effect on our business and could potentially cause us to cease operations.
We may not be successful in our efforts to further develop our current product candidates. We are not permitted to market or promote any of our product candidates before we receive regulatory approval from the FDA or comparable foreign regulatory authorities, and we may never receive such regulatory approval for any of our product candidates. Each of our product candidates is in the early stages of development and will require significant additional clinical development, management of preclinical, clinical, and manufacturing activities, regulatory approval, adequate manufacturing supply, a commercial organization, and significant marketing efforts before we generate any revenue from product sales, if at all.
We have never completed a product development program in neurodegeneration. None of our product candidates in neurodegeneration have advanced into late-stage development and it may be years before any such study is initiated, if at all. Further, we cannot be certain that any of our product candidates will be successful in clinical studies. We may in the future advance product candidates into clinical studies and terminate such studies prior to their completion.
If any of our product candidates successfully complete clinical studies, we may seek regulatory approval to market our product candidates in the U.S., the European Union, and in additional foreign countries where we believe there is a viable commercial opportunity. We may never receive regulatory approval to market any product candidates even if such product candidates successfully complete clinical studies, which would adversely affect our viability. To obtain regulatory approval
28
in countries outside the U.S., we would need to comply with numerous and varying regulatory requirements of such other countries regarding safety, efficacy, manufacturing and controls, clinical studies, commercial sales, pricing, and distribution of our product candidates. Even if we are successful in obtaining approval in one jurisdiction, we cannot ensure that we will obtain approval in any other jurisdictions. If we are unable to obtain approval for our product candidates in multiple jurisdictions, our business, financial condition, results of operations, and our growth prospects could be negatively affected.
Even if we receive regulatory approval to market any of our product candidates, whether for the treatment or diagnosis of neurodegenerative diseases or other diseases, we cannot provide assurance that any such product candidate will be successfully commercialized, widely accepted in the marketplace or more effective than other commercially available alternatives.
Investment in biopharmaceutical product development involves significant risk that any product candidate will fail to demonstrate adequate efficacy or an acceptable safety profile, gain regulatory approval, and become commercially viable. We cannot provide any assurance that we will be able to successfully advance any of our product candidates through the development process or, if approved, successfully commercialize any of our product candidates.
Alzheimer’s disease has failed every attempt at drug approval.
Despite billions of dollars invested by the biopharmaceutical industry in research programs to develop novel therapeutics for Alzheimer’s disease, no FDA approved treatments have been developed in the past 15 years. In that span of time, many new types and classes of drugs have been developed and tested in Alzheimer’s disease, including monoclonal antibodies, g-secretase modulators and inhibitors, β-site amyloid precursor protein cleaving enzyme (BACE) inhibitors, receptor for advanced glycation end-products (RAGE) inhibitors, nicotinic agonists, serotonin subtype receptor (5HT6) antagonists, and others. All of these scientific programs have failed in clinical testing.
We may not be successful in our efforts to expand our technology or product candidates in other indications.
Our drug development strategy is to clinically test and seek regulatory approval for our product candidates in Alzheimer’s disease, our primary indication. We may expand our research efforts outside of this primary indication and into other areas of clinical medicine based on genetic, biological or mechanistic overlap with the primary indication. Conducting clinical studies for additional indications for our product candidates will require substantial technical, financial and human resources and is prone to the inherent risks of failure in drug development. We cannot provide you any assurance that we will be successful in our effort to expand our technology or our product candidates in additional indications, even if we obtain approval for our product candidate in Alzheimer’s disease.
We only have two product candidates in clinical development and we may not be successful in our efforts to continue to create additional product candidates. If we fail to successfully identify and develop additional product candidates, our commercial opportunity may be limited.
Identifying, developing, obtaining regulatory approval, and commercializing additional product candidates requires substantial expertise and funding and is prone to the risks of failure inherent in drug development. We cannot provide any assurance that we will be able to successfully identify or acquire additional product candidates, advance any additional product candidates through the development process, or assemble sufficient resources to identify, acquire, or develop additional product candidates. If we are unable to successfully identify, acquire, develop, and commercialize additional product candidates, our commercial opportunity may be limited.
Early indications of safety, tolerability or biomarker results from our small clinical studies with PTI-125 may not predict the results of later studies
Results of a Phase 1 clinical study with PTI-125 demonstrated safety, tolerability and pharmacokinetics in 24 healthy subjects exposed to 50-200 mg in a single ascending dose study. However, this was a small, “first-in-human” Phase 1 study designed to assess the initial safety characteristics of PTI-125 in healthy subjects and this study was not designed to, and did not, evaluate safety, tolerability and efficacy of PTI-125 in patients. Additional large, well-controlled, multi-dose studies will be required to evaluate the safety, tolerability and efficacy of PTI-125 to treat patients with any indication, including Alzheimer’s disease. There can be no assurance that such future studies will demonstrate the safety, tolerability or efficacy of PTI-125.
29
Results of a Phase 2a clinical study with PTI-125 demonstrated a reduction in biomarkers of disease. However, this was a small, first-in-patient, clinical-proof-of-concept Phase 2a study designed to assess the initial safety characteristics of PTI-125 in patients. Our Phase 2a study was not designed to, and did not, evaluate large-scale or long-term safety, tolerability and efficacy of PTI-125 in patients. Additional large, well-controlled, multi-dose studies will be required to evaluate the safety, tolerability and efficacy of PTI-125 to treat patients with any indication, including Alzheimer’s disease. There can be no assurance that such future studies will demonstrate the safety, tolerability or efficacy of PTI-125. The failure of PTI-125 to show safety, tolerability or efficacy in any future clinical studies would significantly harm our business.
We have never obtained FDA approval for a diagnostic test and we may not be able to secure such approval in a timely manner or at all.
We are developing a blood-based diagnostic test for Alzheimer’s disease, called SavaDx, which will require FDA approval prior to commercialization. Our diagnostic product candidate, marketing, sales and development activities and manufacturing processes are subject to extensive and rigorous regulation by the FDA pursuant to the FDCA, by comparable agencies in foreign countries, and by other regulatory agencies and governing bodies. Under the FDCA, a diagnostic must receive FDA clearance or approval before it can be commercially marketed in the U.S. The process of obtaining marketing approval or clearance from the FDA or by comparable agencies in foreign countries for new products could:
|
· |
take a significant period of time; |
|
· |
require the expenditure of substantial resources; |
|
· |
involve rigorous pre-clinical testing, as well as increased post-market surveillance; |
|
· |
require changes to products; and |
|
· |
result in limitations on the indicated uses of products. |
If we do not compete effectively with scientific and commercial competitors, we may not be able to successfully develop our diagnostic test for Alzheimer’s disease.
The field of clinical laboratory testing is highly competitive. Diagnostic tests that are developed are characterized by rapid technological change. Our competitors in the U.S. and abroad are numerous and include, among others, major diagnostic companies, reference laboratories, molecular diagnostic firms, universities and other research institutions. Most of our potential competitors have considerably greater financial, technical, marketing and other resources than we do, which may allow these competitors to discover important biological markers and determine their function before we do. We could be adversely affected if we do not discover proteins or biomarkers and characterize their function, develop diagnostic and pharmaceutical and clinical services based on these discoveries, obtain required regulatory and other approvals and launch these tests and their related services before our competitors. We also expect to encounter significant competition with respect to any diagnostic tests that we may develop or commercialize. Those companies that bring to market new diagnostic tests before we do may achieve a significant competitive advantage in marketing and commercializing their tests. We may not be able to develop additional diagnostic tests successfully and we may not obtain or enforce patents covering these tests that provide protection against our competitors. Moreover, our competitors may succeed in developing diagnostic tests that circumvent our technologies or tests. Furthermore, our competitors may succeed in developing technologies or tests that are more effective or less costly than those developed by us or that would render our technologies or tests less competitive or obsolete. We expect competition to intensify in the fields in which we are involved as technical advances in these fields occur and become more widely known and changes in intellectual property laws generate challenges to our intellectual property position.
Our blood-based diagnostic to detect Alzheimer’s disease, called SavaDx, relies in part on the use of research antibodies that we source from commercial vendors. Research antibodies are not consistently available or reliable, and we will need to develop our own proprietary antibodies to advance our diagnostic program.
SavaDx currently relies on the use of commercially available antibodies, which are complex molecules that can recognize and bind to an intended protein. Commercially available antibodies present certain technical flaws, such as improper
30
validation, significant batch-to-batch variations or inconsistent storage, any of which can jeopardize our studies and experiments. Because antibody underperformance can be a significant drain on time and resources, we are developing and validating our own, fit-for-purpose antibody for use with SavaDx and such technical activities are on-going. The complexity of developing our own antibody gives rise to many technical issues that are challenging to solve, and we cannot be certain that we will be able to successfully complete any of these activities, in which case our program may be harmed.
We are heavily dependent on the success of PTI-125 and SavaDx, our product candidates which are still under clinical development. If these product candidates do not receive regulatory approval, our business may be harmed.
Since inception, we have not succeeded in getting regulatory approval for our product candidates and we may never do so. In recent years we have invested a significant portion of our efforts and financial resources in the development of PTI-125 and SavaDx for the treatment and detection of Alzheimer’s disease, respectively. Our future success is substantially dependent on our ability to successfully complete clinical development and obtain regulatory approval for our product candidates, which may never occur. We expect that a substantial portion of our efforts and expenditures over the next few years will be devoted to PTI-125 and SavaDx. This will require additional clinical development, management of clinical and manufacturing activities, regulatory approval in one or more national jurisdictions and obtaining commercial-scale manufacturing supply. Substantial investment and significant efforts will be required before we can generate any revenues from any commercial sales. We cannot be certain that we will be able to successfully complete any of these activities.
We have concentrated a substantial portion of our research and development efforts on the treatment and detection of Alzheimer’s disease, an area of research that has seen significant failure rates. Further, our product candidates are based on new scientific approaches and novel technology, which makes it difficult to predict the time and cost of product candidate development.
We focus substantially all of our research and development efforts on addressing neurodegenerations, such as Alzheimer’s disease. Collectively, efforts by biopharmaceutical companies in the field of neurodegenerative diseases have seen many failures and limited success in drug development. For example, there are no therapeutic options available to reverse Alzheimer’s disease, or even to halt its progress. Our future success is highly dependent on the successful development of our product candidates for treating Alzheimer’s disease. Developing and, if approved, commercializing our product candidates for treatment of Alzheimer’s disease subjects us to many challenges, including obtaining regulatory approval from the FDA and other regulatory authorities who have only a limited set of precedents to rely on. We cannot be sure that our approach will yield satisfactory therapeutic products that are safe and effective, scalable, or profitable.
Our Phase 2 clinical studies with PTI-125 in patients with Alzheimer’s disease are not designed to show a statistically meaningful difference between those patients who receive placebo and those who receive drug.
Clinical research data is often analyzed with statistical probability (p-value) to address the question of whether a clinical observation is related to a treatment effect, a random effect or something else. This, in turn, requires a clinical study to incorporate a sufficiently large sample patient population to infer the appropriate statistical analysis. By design, our Phase 2 clinical program with PTI-125 does not include a sufficiently large patient population to generate statistical probability on clinical observations. This feature may make it difficult for investors to properly interpret whether clinical observations in our Phase 2 studies with PTI-125, if any, are important or meaningful. Conversely, our clinical studies may generate statistically meaningful data (i.e., p<0.05) with regard to biomarkers, or other endpoints, that has trivial or no clinical importance. In general, the distinction between statistical significance and clinical significance is a complex area of research that continues to evolve and may be subject to differences of opinion among scientists, clinicians and other professionals.
We may encounter substantial delays in our clinical studies or may not be able to conduct or complete our clinical studies on the timelines we expect, if at all.
Clinical testing is expensive, time consuming, and subject to uncertainty. We cannot guarantee that any clinical studies will be conducted as planned or completed on schedule, if at all. We cannot be sure that submission of an IND or a clinical study application (CTA) will result in the FDA or European Medicines Agency (EMA), as applicable, allowing clinical studies to begin in a timely manner, if at all. Moreover, even if these studies begin, safety or other issues may arise that could suspend or terminate such clinical studies. A failure of one or more clinical studies can occur at any stage of testing, and our future clinical studies may not be successful. Events that may prevent successful or timely initiation or completion of clinical studies include:
31
|
· |
inability to generate sufficient preclinical, toxicology, or other in vivo or in vitro data to support the initiation or continuation of clinical studies; |
|
· |
delays in confirming target engagement, patient selection, or other relevant biomarkers to be utilized in preclinical and clinical product candidate development; |
|
· |
delays in reaching a consensus with regulatory agencies on study design; |
|
· |
delays in reaching an agreement on acceptable terms with prospective contract research organizations (CROs) and clinical study sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and clinical study sites; |
|
· |
delays in identifying, recruiting, and training suitable clinical investigators; |
|
· |
delays in obtaining required IRB approval for each clinical study site; |
|
· |
imposition of a temporary or permanent clinical hold by regulatory agencies for a number of reasons, including: |
|
· |
after review of an IND or amendment, CTA or amendment, or equivalent application or amendment; |
|
· |
as a result of a new safety finding that presents unreasonable risk to clinical study participants; |
|
· |
a negative finding from an inspection of our clinical study operations or study sites; or |
|
· |
the finding that the investigational protocol or plan is clearly deficient to meet its stated objectives; |
|
· |
delays in identifying, recruiting, and enrolling suitable patients to participate in our clinical studies, and delays caused by patients withdrawing from clinical studies, or failing to return for post-treatment follow-up; |
|
· |
delays caused by disease epidemics or pandemics, such as COVID-19, a novel coronavirus first detected in 2019 and for which no specific vaccine or treatment are currently available; |
|
· |
difficulty collaborating with patient groups and investigators; |
|
· |
failure by our CROs, other third parties, or us to adhere to clinical study requirements; |
|
· |
failure to perform in accordance with the FDA’s or any other regulatory authority’s Code of Good Clinical Practice (GCPs) requirements, or applicable EMA or other regulatory guidelines in other countries; |
|
· |
occurrence of adverse events associated with the product candidate that are viewed to outweigh its potential benefits; |
|
· |
changes in regulatory requirements and guidance that require amending or submitting new clinical protocols; |
|
· |
changes in the standard of care on which a clinical development plan was based, which may require new or additional studies; |
|
· |
the cost of clinical studies of our product candidates being greater than we anticipate; |
|
· |
clinical studies of our product candidates producing negative or inconclusive results, which may result in our deciding, or regulators requiring us, to conduct additional clinical studies or abandon product development programs; and |
32
|
· |
delays in manufacturing, testing, releasing, validating, or importing/exporting sufficient stable quantities of our product candidates for use in clinical studies or the inability to do any of the foregoing. |
Any inability to successfully initiate or complete clinical studies could result in additional costs to us or impair our ability to generate revenue. In addition, if we make manufacturing or formulation changes to our product candidates, we may be required to, or we may elect, to conduct additional studies to bridge our modified product candidates to earlier versions. Clinical study delays could also shorten any periods during which our products have patent protection and may allow our competitors to bring products to market before we do, which could impair our ability to successfully commercialize our product candidates and may harm our business and results of operations.
We could also encounter delays if a clinical study is suspended or terminated by us, by the data safety monitoring board for such study or by the FDA, EMA, or any other regulatory authority, or if the IRBs of the institutions in which such studies are being conducted suspend or terminate the participation of their clinical investigators. Such authorities may suspend or terminate a clinical study due to a number of factors, including failure to conduct the clinical study in accordance with regulatory requirements or our clinical protocols, inspection of the clinical study operations or study site by the FDA, EMA, or other regulatory authorities resulting in the imposition of a clinical hold, unforeseen safety issues or adverse side effects, failure to demonstrate a benefit from using a product candidate, changes in governmental regulations or administrative actions, or lack of adequate funding to continue the clinical trial.
We may in the future advance product candidates into clinical studies and terminate such studies prior to their completion, which could adversely affect our business.
Delays in the completion of any clinical study of our product candidates will increase our costs, slow down our product candidate development and approval process and delay, or potentially jeopardize our ability to commence product sales and generate revenue. In addition, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical studies may also ultimately lead to the denial of regulatory approval of our product candidates.
We may encounter difficulties enrolling patients in our clinical studies, and our clinical development activities could thereby be delayed or otherwise adversely affected.
The timely completion of clinical studies in accordance with their protocols depends, among other things, on our ability to enroll enough patients who remain in the study until its conclusion. We may experience difficulties in patient enrollment in our clinical studies for a variety of reasons, including:
|
· |
the size and nature of the patient population; |
|
· |
the patient eligibility criteria defined in the protocol, including biomarker-driven identification and/or certain highly-specific criteria related to stage of disease progression, which may limit the patient populations eligible for our clinical studies to a greater extent than competing clinical studies for the same indication that do not have biomarker-driven patient eligibility criteria; |
|
· |
the size of the study population required for analysis of the trial’s primary endpoints; |
|
· |
the design of the study; |
|
· |
our ability to recruit clinical study investigators with the appropriate competencies and experience; |
|
· |
competing clinical studies for similar therapies or targeting patient populations meeting our patient eligibility criteria; |
|
· |
clinicians’ and patients’ perceptions as to the potential advantages and side effects of the product candidate being studied in relation to other available therapies and product candidates; |
33
|
· |
our ability to obtain and maintain patient consents; and |
|
· |
the risk that patients enrolled in clinical studies will not complete such studies, for any reason. |
Our clinical studies may fail to demonstrate substantial evidence of the safety and efficacy of our product candidates, which would prevent, delay, or limit the scope of regulatory approval and commercialization.
Before obtaining regulatory approvals for any of our product candidates, we must demonstrate through lengthy, complex, and expensive preclinical experiments and clinical studies that our product candidates are both safe and effective for use in an intended population. Each product candidate must demonstrate an adequate risk versus benefit profile in its intended patient population and for its intended use.
Clinical testing is expensive and can take many years to complete, and its outcome is inherently uncertain. Failure can occur at any time during the clinical study process. The results of preclinical studies of our product candidates may not be predictive of the results of early-stage or later-stage clinical studies, and results of early clinical studies of our product candidates may not be predictive of the results of later-stage clinical studies. The results of clinical studies in one set of patients or disease indications may not be predictive of those obtained in another. In some instances, there can be significant variability in safety or efficacy results between different clinical studies of the same product candidate due to numerous factors, including changes in study procedures set forth in protocols, differences in the size and type of the patient populations, changes in and adherence to the dosing regimen, and other clinical study protocols and the rate of dropout among clinical study participants. Open-label extension studies may also extend the timing and cost of a clinical study substantially. Product candidates in later stages of clinical studies may fail to show the desired safety and efficacy profile despite having progressed through preclinical studies and initial clinical studies. Many companies in the biopharmaceutical industry have suffered significant setbacks in advanced clinical studies due to lack of efficacy or unacceptable safety issues, notwithstanding promising results in earlier studies. This is particularly true in neurodegenerative diseases, where failure rates historically have been higher than in many other disease areas. Most product candidates that begin clinical studies are never approved by regulatory authorities for commercialization.
We have limited experience in designing clinical studies in neurodegeneration and may be unable to design and execute a clinical study to support marketing approval. We cannot be certain that our current clinical studies or any other future clinical studies will be successful. Additionally, any safety concerns observed in any one of our clinical studies in our targeted indications could limit the prospects for regulatory approval of our product candidates in those and other indications, which could have a material adverse effect on our business, financial condition, and results of operations.
In addition, even if such clinical studies are successfully completed, we cannot guarantee that the FDA or foreign regulatory authorities will interpret the results as we do, and more studies could be required before we submit our product candidates for approval. To the extent that the results of the studies are not satisfactory to the FDA or foreign regulatory authorities for support of a marketing application, we may be required to expend significant resources, which may not be available to us, to conduct additional studies in support of potential approval of our product candidates. Even if regulatory approval is secured for any of our product candidates, the terms of such approval may limit the scope and use of our product candidates, which may also limit its commercial potential.
The market opportunities for PTI-125 and SavaDx, if approved, may be smaller than we anticipate.
If our clinical development programs succeed, we expect to seek regulatory approval of PTI-125 and SavaDx for patients with Alzheimer’s disease. Our projections of the number of patients with Alzheimer’s disease is based on our beliefs and estimates. These estimates have been derived from a variety of sources, including scientific literature, patient foundations and market research, and may prove to be incorrect. The actual number of patients may turn out to be lower than expected. Additionally, the potential patient population for our current programs or future product candidates may be limited. Even if we obtain significant market share for any product candidate, if approved, if the potential target populations are smaller than anticipated, we may never achieve profitability without obtaining marketing approval for additional indications.
34
We face significant competition in an environment of rapid technological and scientific change, and there is a possibility that our competitors may achieve regulatory approval before us or develop therapies that are safer, more advanced, or more effective than ours, any of which may harm our business operations.
The development and commercialization of new product candidates is highly competitive. Moreover, the neurodegenerative field is characterized by intense and increasing competition, and a strong emphasis on intellectual property. We may face competition with respect to any of our product candidates that we seek to develop or commercialize in the future from major pharmaceutical companies, specialty pharmaceutical companies, and biotechnology companies worldwide. Potential competitors also include academic institutions, government agencies, and other public and private research organizations that conduct research, seek patent protection, and establish collaborative arrangements for research, development, manufacturing, and commercialization.
Several large pharmaceutical and biotechnology companies are currently pursuing the development of products for the treatment of neurodegenerative diseases, including Alzheimer’s disease. Many of these current or potential competitors, either alone or with their strategic partners, have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical studies, obtaining regulatory approvals, and marketing approved products than we do.
Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient, or are less expensive than any products that we may develop. Furthermore, currently approved products could be discovered to have application for treatment of neurodegenerative disease indications, which could give such products significant advantages over any of our product candidates. Our competitors also may obtain FDA, EMA, or other regulatory approval for their products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market. Additionally, products or technologies developed by our competitors may render our potential product candidates uneconomical or obsolete, and we may not be successful in marketing any product candidates we may develop against competitors.
In addition, we could face litigation or other proceedings with respect to the scope, ownership, validity, and/or enforceability of our patents relating to our competitors’ products and our competitors may allege that our products infringe, misappropriate, or otherwise violate their intellectual property. The availability of our competitors’ products could limit the demand, and the price we are able to charge, for any products that we may develop and commercialize.
The manufacture of our product candidates is complex, and we may encounter difficulties in production. If we or any of our third-party manufacturers encounter such difficulties, or fail to meet rigorously enforced regulatory standards, our ability to provide supply of our product candidates for clinical studies or our products for patients, if approved, could be delayed or stopped, or we may be unable to maintain a commercially viable cost structure.
The processes involved in manufacturing our product candidates are complex, expensive, highly regulated, and subject to multiple risks. Further, as product candidates are developed through preclinical studies to late-stage clinical studies towards approval and commercialization, it is common that various aspects of the development program, such as manufacturing methods, are altered along the way in an effort to optimize processes and results. Such changes carry the risk that they will not achieve these intended objectives, and any of these changes could cause our product candidates to perform differently and affect the results of planned clinical studies or other future clinical studies.
In order to conduct clinical studies of our product candidates, or supply commercial products, if approved, we will need to manufacture them in large quantities. Our contract development and manufacturing organizations (CDMOs) may be unable to successfully increase the manufacturing capacity for any of our product candidates in a timely or cost-effective manner, or at all. In addition, quality issues may arise during scale-up activities. If our CDMOs are unable to successfully scale up the manufacture of our product candidates in sufficient quality and quantity, the development, testing, and clinical studies of that product candidate may be delayed or become infeasible, and regulatory approval or commercial launch of any resulting product may be delayed or not obtained, which could significantly harm our business. The same risk would apply to our internal manufacturing facilities, should we in the future decide to build internal manufacturing capacity. In addition, building internal manufacturing capacity would carry significant risks in terms of being able to plan, design, and execute on a complex project to build manufacturing facilities in a timely and cost-efficient manner.
35
In addition, the manufacturing process for any products that we may develop is subject to FDA, EMA, and foreign regulatory authority approval processes, and continuous oversight, and we will need to contract with manufacturers who can meet all applicable FDA, EMA, and foreign regulatory authority requirements, including complying with cGMP on an ongoing basis. If we or our third-party manufacturers are unable to reliably produce products to specifications acceptable to the FDA, EMA, or other regulatory authorities, we may not obtain or maintain the approvals we need to commercialize such products. Even if we obtain regulatory approval for any of our product candidates, there is no assurance that either we or our CDMOs will be able to manufacture the approved product to specifications acceptable to the FDA, EMA, or other regulatory authorities, to produce it in sufficient quantities to meet the requirements for the potential launch of the product, or to meet potential future demand. Any of these challenges could delay completion of clinical studies, require bridging clinical studies or the repetition of one or more clinical studies, increase clinical study costs, delay approval of our product candidates, impair commercialization efforts, increase our cost of goods, and have an adverse effect on our business, financial condition, results of operations, and growth prospects.
If our product candidates receive regulatory approval, we and our collaborators will be subject to ongoing FDA obligations and continued regulatory review, such as continued safety reporting requirements, and we and our collaborators may also be subject to additional FDA post-marketing obligations or new regulations, all of which may result in significant expense and limit our and our collaborators’ ability to commercialize our potential drugs.
Any regulatory approvals that our product candidates receive may also be subject to limitations on the indicated uses for which the drug may be marketed or contain requirements for potentially costly post-marketing follow-up studies. In addition, if the FDA approves any of our product candidates, the labeling, packaging, adverse event reporting, storage, advertising, promotion and record keeping for the drug will be subject to extensive regulatory requirements. The subsequent discovery of previously unknown problems with the drug, including but not limited to adverse events of unanticipated severity or frequency, or the discovery that adverse events previously observed in preclinical research or clinical studies that were believed to be minor actually constitute much more serious problems, may result in restrictions on the marketing of the drug, and could include withdrawal of the drug from the market.
The FDA’s policies may change, and additional government regulations may be enacted that could prevent or delay regulatory approval of our product candidates. We cannot predict the likelihood, nature or extent of adverse government regulation that may arise from future legislation or administrative action, either in the U.S. or abroad. If we are not able to maintain regulatory compliance, we may be subject to fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution. Any of these events could prevent us from marketing our drugs and our business could suffer.
Risks Related to Regulatory Approval and Other Legal Compliance Matters
The regulatory cycles of the FDA, EMA, and comparable foreign regulatory authorities are lengthy, time consuming, and inherently unpredictable. If we are ultimately unable to obtain regulatory approval for our product candidates, we will be unable to generate product revenue and our business will be substantially harmed.
The time required to obtain approval by the FDA, EMA, and comparable foreign regulatory authorities is unpredictable, typically takes many years following the commencement of clinical studies, and depends upon numerous factors, including the type, complexity, and novelty of the product candidates involved. In addition, approval policies, regulations, or the type and amount of clinical data necessary to gain approval may change during the course of a product candidate’s clinical development and may vary among jurisdictions, which may cause delays in the approval or the decision not to approve an application. Regulatory authorities have substantial discretion in the approval process and may refuse to accept any application or may decide that our data are insufficient for approval and require additional preclinical, clinical, or other studies. The FDA has not approved a new drug for Alzheimer’s disease since 2003. We have not obtained regulatory approval for any product candidate, including our product candidates aimed at Alzheimer’s disease, and it is possible that none of our existing product candidates or any product candidates we may seek to develop in the future will ever obtain regulatory approval.
Applications for our product candidates could fail to receive regulatory approval in an initial or subsequent indication for many reasons, including but not limited to the following:
36
|
· |
the FDA, EMA, or comparable foreign regulatory authorities may disagree with the design, implementation, or results of our clinical studies; |
|
· |
the FDA, EMA, or comparable foreign regulatory authorities may determine that our product candidates are not safe and effective, only moderately effective or have undesirable or unintended side effects, toxicities, or other characteristics that preclude our obtaining marketing approval or prevent or limit commercial use; |
|
· |
the population studied in the clinical program may not be sufficiently broad or representative to assure efficacy and safety in the full population for which we seek approval; |
|
· |
we may be unable to demonstrate to the FDA, EMA or comparable foreign regulatory authorities that a product candidate’s risk-benefit ratio when compared to the standard of care is acceptable; |
|
· |
the FDA, EMA, or comparable foreign regulatory authorities may disagree with our interpretation of data from preclinical studies or clinical studies; |
|
· |
the data collected from clinical studies of our product candidates may not be sufficient to support the submission of a new drug application (NDA), or other submission or to obtain regulatory approval in the United States or elsewhere; |
|
· |
the FDA, EMA, or comparable foreign regulatory authorities may fail to approve the manufacturing processes, test procedures, and specifications, or facilities of third-party manufacturers with which we contract for clinical and commercial supplies; and |
|
· |
the approval policies or regulations of the FDA, EMA, or comparable foreign regulatory authorities may significantly change in a manner rendering our clinical data insufficient for approval. |
This lengthy approval process, as well as the unpredictability of the results of clinical studies, may result in our failing to obtain regulatory approval to market any of our product candidates, which would significantly harm our business, results of operations, and growth prospects.
Our product candidates may cause undesirable side effects or have other properties that could halt their clinical development, prevent their regulatory approval, limit their commercial potential, or result in significant negative consequences.
We have not yet completed long term safety studies with PTI-125 to determine if this product candidate is safe for humans. Adverse events or other undesirable side effects caused by PTI-125 could cause us or regulatory authorities to interrupt, delay, or halt clinical studies and could result in a more restrictive label or the delay or denial of regulatory approval by the FDA, EMA, or other comparable foreign regulatory authorities.
Drug-related side effects could affect patient recruitment, the ability of enrolled patients to complete the study, and/or result in potential product liability claims. We are required to maintain product liability insurance pursuant to certain of our development and commercialization agreements. We may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses due to liability. A successful product liability claim or series of claims brought against us could adversely affect our results of operations, business, and reputation. In addition, regardless of merit or eventual outcome, product liability claims may result in impairment of our business reputation, withdrawal of clinical study participants, costs due to related litigation, distraction of management’s attention from our primary business, initiation of investigations by regulators, substantial monetary awards to patients or other claimants, the inability to commercialize our product candidates, and decreased demand for our product candidates, if approved for commercial sale.
Our employees, independent contractors, consultants, commercial partners, and vendors may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements.
We are exposed to the risk of fraud, misconduct, or other illegal activity by our employees, independent contractors, consultants, commercial partners, and vendors. Misconduct by these parties could include intentional, reckless, and negligent conduct that fails to:
37
|
· |
comply with the laws of the FDA, EMA, and other comparable foreign regulatory authorities; |
|
· |
provide true, complete, and accurate information to the FDA, EMA, and other comparable foreign regulatory authorities; |
|
· |
comply with manufacturing standards we have established; |
|
· |
comply with healthcare fraud and abuse laws in the U.S. and similar foreign fraudulent misconduct laws; or |
|
· |
report financial information or data accurately or to disclose unauthorized activities to us. |
Activities subject to laws also involve the improper use of information obtained in the course of patient recruitment for clinical studies, which could result in regulatory sanctions and cause serious harm to our reputation. Further, it is not always possible to identify and deter misconduct by employees and third parties, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
A recent federal court ruling may mandate significant new disclosure requirements for clinical data dating back over a decade. In many cases, we were the responsible party for generating such clinical data, but it may be difficult or not feasible to access such data, which may leave us in a conflicting position in regards to these new disclosure requirements.
A recent federal court ruling may require all clinical study sponsors to report a decade’s worth of previously exempted clinical study data to the federal government for publication on ClinicalTrials.gov. In February 2020, the U.S. District Court for the Southern District of New York invalidated a prior interpretation of NIH regulations that had exempted many clinical studies conducted between 2007 and 2017 from reporting requirements mandated by the Food and Drug Administration Amendments Act. If this court ruling takes effect without appeal, or if it is upheld on appeal, it could require us to submit an onerous amount of old clinical data to the federal government. In many cases, we were the responsible party for generating such clinical data, but such prior data may be difficult or not feasible for us to access as a result of our strategic shift away from analgesic drug development in 2019. We may no longer have control over, or access to, prior clinical data that we may legally be required to report to NIH in the future. Furthermore, it is unclear whether such new disclosure requirements apply to inactive, failed or abandoned drug development programs. As a result of these uncertainties, the government’s recent ruling may leave us in a conflicted position or out of compliance with new disclosure requirements. We currently do not and cannot understand or anticipate the full impact and potential implications of this recent court ruling on our business.
Risks Related to Our Reliance on Third Parties
We expect to rely on third parties to conduct our studies and some aspects of our research, and such third parties may not perform satisfactorily, including failing to meet deadlines for the completion of such studies, research, or testing.
We currently rely and expect to continue to rely on third parties, such as CROs, clinical data management organizations, medical institutions, and clinical investigators, to conduct some aspects of our research and preclinical testing and our clinical studies. Any of these third parties may terminate their engagements with us or be unable to fulfill their contractual obligations. If we need to enter into alternative arrangements, it will delay our product development activities.
Our reliance on these third parties for research and development activities reduces our control over these activities but does not relieve us of our responsibilities. For example, we remain responsible for ensuring that all of our clinical studies are conducted in accordance with the general investigational plan and protocols for the trial. Moreover, the FDA requires us to comply with the norms of Good Clinical Practice (GCPs) for conducting, recording, and reporting the results of clinical studies to assure that data and reported results are credible, reproducible, and accurate and that the rights, integrity, and confidentiality of study participants are protected. We also are required to register ongoing clinical studies and post the results of completed clinical studies on a government-sponsored database within certain timeframes. Failure to do so can result in fines, adverse publicity, and civil and criminal sanctions.
38
If our third party vendors do not successfully carry out their contractual duties, meet expected deadlines, or conduct studies in accordance with regulatory requirements or our stated protocols, we will not be able to obtain, or may be delayed in obtaining, marketing approvals for any product candidates we may develop and will not be able to, or may be delayed in our efforts to, successfully commercialize our product candidates. For example, one of our vendors failed to fully comply with certain Good Laboratory Practice (GLP) norms in its research facility, which required us to repeat a lab study at a different research site.
We also expect to rely on other third parties to store and distribute drug supplies for our clinical studies. Any performance failure on the part of our distributors, including with the shipment of any drug supplies, could delay clinical development or marketing approval of any product candidates we may develop or commercialization of our product candidates, producing additional losses and depriving us of potential product revenue.
We contract with third parties for the manufacture of materials for our research programs, preclinical studies, clinical studies, and for commercialization of any product candidates that we may develop. This reliance on third parties carries and may increase the risk that we will not have sufficient quantities of such materials, product candidates, or any product candidates that we may develop and commercialize, or that such supply will not be available to us at an acceptable cost, which could delay, prevent, or impair our development or commercialization efforts.
We do not have any manufacturing facilities. We currently rely on CDMOs for all of the manufacture of our materials for preclinical studies and clinical studies and expect to continue to do so for preclinical studies, clinical studies, and for commercial supply of any product candidates that we may develop. We currently have established relationships with several CDMOs for the manufacturing of our product candidates. We may be unable to establish any further agreements with CDMOs or to do so on acceptable terms. Even if we are able to establish agreements with third-party manufacturers, reliance on CDMOs entails additional risks, including:
|
· |
the possible breach of the manufacturing agreement by the third party; |
|
· |
the possible termination or nonrenewal of the agreement by the third party at a time that is costly or inconvenient for us; |
|
· |
reliance on the third party for regulatory compliance, quality assurance, safety, and pharmacovigilance and related reporting; and |
|
· |
the inability to produce required volume in a timely manner and to quality standards. |
Third-party manufacturers may not be able to comply with cGMP regulations or similar regulatory requirements outside the U.S. Our failure, or the failure of our CDMOs, to comply with applicable regulations could result in clinical holds on our studies, sanctions being imposed on us, including fines, injunctions, civil penalties, delays, suspension or withdrawal of approvals, seizures, or recalls of product candidates or product candidates, operating restrictions, and criminal prosecutions, any of which could significantly and adversely affect supplies of our product candidates and harm our business, financial condition, results of operations, and growth prospects.
Any product candidates that we may develop may compete with other product candidates and products for access to manufacturing facilities. There are a limited number of manufacturers that operate under cGMP regulations and that might be capable of manufacturing for us.
Any performance failure on the part of our existing or future third-party manufacturers could delay clinical development or marketing approval. If any one of our current contract manufacturers cannot perform as agreed, we may be required to replace that manufacturer and may incur added costs and delays in identifying and qualifying any such replacement. Furthermore, securing and reserving production capacity with contract manufacturers may result in significant costs.
Our current and anticipated future dependence upon others for the manufacture of any product candidates we may develop may adversely affect our future profit margins and our ability to commercialize any medicines that receive marketing approval on a timely and competitive basis.
39
We depend on third-party suppliers for key raw materials used in our manufacturing processes, and the loss of these third-party suppliers or their inability to supply us with adequate raw materials could harm our business.
We rely on third-party suppliers for the supply of the raw materials required for the production of our product candidates, and we expect to continue to rely on third party manufacturers for the commercial supply of any of our product candidates for which we obtain marketing approval. Our dependence on these third-party suppliers and the challenges we may face in obtaining adequate supplies of raw materials involve several risks, including limited control over pricing, availability, quality, and delivery schedules. As a small company, our negotiation leverage is limited and we are likely to get lower priority than our competitors who are larger than we are. We do not have long-term supply agreements, and we purchase our required drug product on a development manufacturing services agreement or purchase order basis. We cannot be certain that our suppliers will continue to provide us with the quantities of these raw materials that we require or satisfy our anticipated specifications and quality requirements. Any supply interruption in limited or sole sourced raw materials could materially harm our ability to manufacture our product candidates until a new source of supply, if any, could be identified and qualified. We may be unable to find a sufficient alternative supply channel in a reasonable time or on commercially reasonable terms. Any performance failure on the part of our suppliers could delay the development and potential commercialization of our product candidates, including limiting supplies necessary for clinical studies and regulatory approvals, which would have a material adverse effect on our business.
Risks Related to Our Intellectual Property
If we are unable to obtain and maintain patent protection for any product candidates we develop, our competitors could develop and commercialize products similar or identical to ours, and our ability to successfully commercialize any product candidates we may develop may be adversely affected.
Our success depends in large part on our ability to obtain and maintain patent protection in the U.S. and other countries with respect to our proprietary product candidates and other technologies we may develop. We seek to protect our proprietary position by filing patent applications in the U.S. and abroad relating to our core programs and product candidates, as well as other technologies that are important to our business. Given that the development of our product candidates is at an early stage, our intellectual property portfolio with respect to certain aspects of our product candidates is also at an early stage. For example, we have filed or intend to file patent applications on aspects of our technology and core product candidates; however, there can be no assurance that any such patent applications will issue as granted patents. Furthermore, in some cases, we have only filed provisional patent applications on certain aspects of our technology and product candidates and each of these provisional patent applications is not eligible to become an issued patent until, among other things, we file a non-provisional patent application within 12 months of the filing date of the applicable provisional patent application. Any failure to file a non-provisional patent application within this timeline could cause us to lose the ability to obtain patent protection for the inventions disclosed in the associated provisional patent applications.
Furthermore, in some cases, we may not be able to obtain issued claims covering compositions relating to our core programs and product candidates, as well as other technologies that are important to our business, and instead may need to rely on filing patent applications with claims covering a method of use and/or method of manufacture for protection of such core programs, product candidates, and other technologies. There can be no assurance that any such patent applications will issue as granted patents, and even if they do issue, such patent claims may be insufficient to prevent third parties, such as our competitors, from utilizing our technology. Any failure to obtain or maintain patent protection with respect to our core programs and product candidates could have a material adverse effect on our business, financial condition, results of operations, and growth prospects.
40
If any of our patent applications do not issue as patents in any jurisdiction, we may not be able to compete effectively.
Changes in either the patent laws or their interpretation in the U.S. and other countries may diminish our ability to protect our inventions, obtain, maintain, and enforce our intellectual property rights and, more generally, could affect the value of our intellectual property or narrow the scope of our patents with respect to our product candidates. With respect to our intellectual property related to our product candidates, we cannot predict whether the patent applications we are currently pursuing will issue as patents in any particular jurisdiction or whether the claims of any issued patents will provide sufficient protection from competitors or other third parties.
The patent prosecution process is expensive, time-consuming, and complex, and we may not be able to file, prosecute, maintain, or enforce all necessary or desirable patent applications at a reasonable cost or in a timely manner. It is also possible that we will fail to identify patentable aspects of our research and development output in time to obtain patent protection. Although we enter into nondisclosure and confidentiality agreements with parties who have access to confidential or patentable aspects of our research and development output, such as our employees, outside scientific collaborators, CROs, CDMOs, consultants, advisors, and other third parties, any of these parties may breach the agreements and disclose such output before a patent application is filed, thereby jeopardizing our ability to seek patent protection. In addition, our ability to obtain and maintain valid and enforceable patents depends on whether the differences between our inventions and the prior art allow our inventions to be patentable. Furthermore, publications of discoveries in the scientific literature often lag behind the actual discoveries, and patent applications in the U.S. and other jurisdictions are typically not published until 18 months after filing, or in some cases not at all. Therefore, we cannot be certain that we were the first to make the inventions claimed in any of our patents or pending patent applications, or that we were the first to file for patent protection of such inventions.
U.S. intellectual property rights around diagnostic methods is a complex, evolving area of law and effective patent claims may not be available to us for our investigational diagnostic product candidate, SavaDx.
The legal system for intellectual property around diagnostic methods is highly complex, remains uncertain and continues to evolve. In the U.S., patent courts have struggled to define a clear means of patent eligibility for modern age diagnostics. Case law interpretations from the U.S. Supreme Court has left certain important scientific advances in the area of diagnostics without effective patent claims. In 2012, the Supreme Court held that a simple process involving correlations between blood test results and patient health is not eligible for patent claims because such processes incorporate “laws of nature”. Since then, different outcomes from different courts, including Federal Circuit, district court and Patent Trial and Appeal Board decisions, have continued to create a sometimes vague or conflicting legal framework for determining the eligibility of patent claims for diagnostic methods. As a result, we cannot be certain how SavaDx fits into the current U.S. legal framework for obtaining effective patent claims. Furthermore, claims for diagnostic methods can be complicated to enforce. For patent infringement to occur with a protected diagnostic, the patented method must generally either be performed by one person in its entirety or performed by multiple parties all under the control or direction of a single party. Accordingly, even if effective patent claims are issued for SavaDx, it may not be practical, possible or even desirable to enforce potential infringement claims around this product candidate.
If the scope of any patent protection we obtain is not sufficiently broad, or if we lose any of our patent protection, our ability to prevent our competitors from commercializing similar or identical technology and product candidates would be adversely affected.
The patent position of biotechnology and pharmaceutical companies generally is highly uncertain, involves complex legal and factual questions, and has been the subject of much litigation in recent years. As a result, the issuance, scope, validity, enforceability, and commercial value of our patent rights are highly uncertain. Our pending and future patent applications may not result in patents being issued which protect our product candidates or other technologies or which effectively prevent others from commercializing competitive technologies and product candidates.
Moreover, the coverage claimed in a patent application can be significantly reduced before the patent is issued, and its scope can be reinterpreted after issuance. Even if patent applications we own currently or in the future issue as patents, they may not issue in a form that will provide us with any meaningful protection, prevent competitors or other third parties from competing with us, or otherwise provide us with any competitive advantage. Any patents to which we have rights may be challenged, narrowed, circumvented, or invalidated by third parties. Consequently, we do not know whether product candidates or other technologies will be protectable or remain protected by valid and enforceable patents. Our competitors or other third parties may be able to circumvent our patents by developing similar or alternative technologies or products in a
41
non-infringing manner which could materially adversely affect our business, financial condition, results of operations and growth prospects.
The issuance of a patent is not conclusive as to its inventorship, scope, validity, or enforceability, and our patents may be challenged in the courts or patent offices in the U.S. and abroad. We may be subject to a third-party pre-issuance submission of prior art to the United States Patent and Trademark Office (USPTO) or become involved in opposition, derivation, revocation, reexamination, post-grant and inter partes review, or interference proceedings or other similar proceedings challenging our patent rights. An adverse determination in any such submission, proceeding, or litigation could reduce the scope of, or invalidate or render unenforceable, such patent rights, allow third parties to commercialize our product candidates or other technologies and compete directly with us, without payment to us, or result in our inability to manufacture or commercialize products without infringing third-party patent rights. Moreover, we may have to participate in interference proceedings declared by the USPTO to determine priority of invention or in post-grant challenge proceedings, such as oppositions in a foreign patent office, that challenge our priority of invention or other features of patentability with respect to our patents and patent applications. Such challenges may result in loss of patent rights, loss of exclusivity, or in patent claims being narrowed, invalidated, or held unenforceable, which could limit our ability to stop others from using or commercializing similar or identical technology and products, or limit the duration of the patent protection of our product candidates and other technologies. Such proceedings also may result in substantial cost and require significant time from our scientists and management, even if the eventual outcome is favorable to us. If we are unsuccessful in any such proceeding or other priority or inventorship dispute, we may be required to obtain and maintain licenses from third parties, including parties involved in any such interference proceedings or other priority or inventorship disputes. Such licenses may not be available on commercially reasonable terms or at all or may be non-exclusive. If we are unable to obtain and maintain such licenses, we may need to cease the development, manufacture, and commercialization of one or more of the product candidates we may develop. The loss of exclusivity or the narrowing of our owned and licensed patent claims could limit our ability to stop others from using or commercializing similar or identical technology and products.
In addition, given the amount of time required for the development, testing, and regulatory review of new product candidates, patents protecting such product candidates might expire before or shortly after such product candidates are commercialized. As a result, our intellectual property may not provide us with sufficient rights to exclude others from commercializing products similar or identical to ours.
We may not be able to protect our intellectual property and proprietary rights throughout the world.
Filing, prosecuting, and defending patents on our product candidates and other technologies in all countries throughout the world would be prohibitively expensive, and the laws of foreign countries may not protect our rights to the same extent as the laws of the U.S.
Consequently, we may not be able to prevent third parties from practicing our inventions in all countries outside the U.S., or from selling or importing products made using our inventions in and into the U.S. or other jurisdictions. Competitors may use our technologies in jurisdictions where we have not obtained patent protection to develop their own products, and, further, may export otherwise infringing products to territories where we have patent protection, but enforcement is not as strong as that in the U.S. These products may compete with our products, and our patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents, trade secrets, and other intellectual property protection, particularly those relating to biotechnology products, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our intellectual property and proprietary rights generally. Proceedings to enforce our intellectual property and proprietary rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our patents at risk of being invalidated or interpreted narrowly, could put our patent applications at risk of not issuing, and could provoke third parties to assert claims against us. We may not prevail in any lawsuits that we initiate, and the damages or other remedies awarded, if any, may not be commercially meaningful. Accordingly, our efforts to enforce our intellectual property and proprietary rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we develop or license.
42
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment, and other requirements imposed by government patent agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees, renewal fees, annuity fees, and various other government fees on patents and applications will be due to be paid to the USPTO and various government patent agencies outside of the U.S. over the lifetime of our owned or licensed patents and applications. The USPTO and various non-U.S. government agencies require compliance with several procedural, documentary, fee payment, and other similar provisions during the patent application process. In some cases, an inadvertent lapse can be cured by payment of a late fee or by other means in accordance with the applicable rules. There are situations, however, in which non-compliance can result in abandonment or lapse of the patent or patent application, resulting in a partial or complete loss of patent rights in the relevant jurisdiction. In such an event, potential competitors might be able to enter the market with similar or identical products or technology, which could have a material adverse effect on our business, financial condition, results of operations, and growth prospects.
Changes in U.S. patent law could diminish the value of patents in general, thereby impairing our ability to protect our products.
Changes in either the patent laws or interpretation of the patent laws in the U.S. could increase the uncertainties and costs surrounding the prosecution of patent applications and the enforcement or defense of issued patents. Assuming that other requirements for patentability are met, prior to March 2013, in the U.S., the first to invent the claimed invention was entitled to the patent, while outside the U.S., the first to file a patent application was entitled to the patent. After March 2013, under the Leahy-Smith America Invents Act (the America Invents Act) enacted in September 2011, the U.S. transitioned to a first inventor to file system in which, assuming that other requirements for patentability are met, the first inventor to file a patent application will be entitled to the patent on an invention regardless of whether a third party was the first to invent the claimed invention. A third party that files a patent application in the USPTO after March 2013, but before us could therefore be awarded a patent covering an invention of ours even if we had made the invention before it was made by such third party. This will require us to be cognizant going forward of the time from invention to filing of a patent application. Since patent applications in the U.S. and most other countries are confidential for a period of time after filing or until issuance, we cannot be certain that we were the first to either (i) file any patent application related to our product candidates or other technologies or (ii) invent any of the inventions claimed in our patents or patent applications.
The America Invents Act also includes a number of significant changes that affect the way patent applications will be prosecuted and also may affect patent litigation. These include allowing third party submission of prior art to the USPTO during patent prosecution and additional procedures to attack the validity of a patent by USPTO administered post-grant proceedings, including post-grant review, inter partes review, and derivation proceedings. Because of a lower evidentiary standard in USPTO proceedings as compared to the evidentiary standard in U.S. federal courts necessary to invalidate a patent claim, a third party could potentially provide evidence in a USPTO proceeding sufficient for the USPTO to hold a claim invalid even though the same evidence would be insufficient to invalidate the claim if first presented in a district court action. Accordingly, a third party may attempt to use the USPTO procedures to invalidate our patent claims that would not have been invalidated if first challenged by the third party as a defendant in a district court action. Therefore, the America Invents Act and its implementation could increase the uncertainties and costs surrounding the prosecution of our patent applications and the enforcement or defense of our issued patents, all of which could have a material adverse effect on our business, financial condition, results of operations, and growth prospects.
In addition, the patent positions of companies in the development and commercialization of pharmaceuticals are particularly uncertain. Recent U.S. Supreme Court rulings have narrowed the scope of patent protection available in certain circumstances and weakened the rights of patent owners in certain situations. This combination of events has created uncertainty with respect to the validity and enforceability of patents, once obtained. Depending on future actions by the U.S. Congress, the federal courts, and the USPTO, the laws and regulations governing patents could change in unpredictable ways that could have a material adverse effect on our existing patent portfolio and our ability to protect and enforce our intellectual property in the future.
43
Issued patents covering our product candidates and other technologies could be found invalid or unenforceable if challenged in court or before administrative bodies in the U.S. or abroad.
If we initiated legal proceedings against a third party to enforce a patent covering our product candidates or other technologies, the defendant could counterclaim that such patent is invalid or unenforceable. In patent litigation in the U.S., defendant counterclaims alleging invalidity or unenforceability are commonplace. Grounds for a validity challenge could be an alleged failure to meet any of several statutory requirements, including lack of novelty, obviousness, or non-enablement. Grounds for an unenforceability assertion could be an allegation that someone connected with prosecution of the patent withheld relevant information from the USPTO, or made a misleading statement, during prosecution. Third parties may raise claims challenging the validity or enforceability of our patents before administrative bodies in the U.S. or abroad, even outside the context of litigation. Such mechanisms include re-examination, post-grant review, inter partes review, interference proceedings, derivation proceedings, and equivalent proceedings in foreign jurisdictions (e.g., opposition proceedings). Such proceedings could result in the revocation of, cancellation of, or amendment to our patents in such a way that they no longer cover our product candidates or other technologies. The outcome following legal assertions of invalidity and unenforceability is unpredictable. With respect to the validity question, for example, we cannot be certain that there is no invalidating prior art, of which we or our licensing partners and the patent examiner were unaware during prosecution. If a third party were to prevail on a legal assertion of invalidity or unenforceability, we would lose at least part, and perhaps all, of the patent protection on our product candidates or other technologies. Such a loss of patent protection would have a material adverse impact on our business, financial condition, results of operations, and growth prospects.
If we do not obtain patent term extension and data exclusivity for any product candidates we may develop, our business may be materially harmed.
Depending upon the timing, duration, and specifics of any FDA marketing approval of any product candidates we may develop, one or more of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984 (Hatch-Waxman Act). The Hatch-Waxman Act permits a patent term extension of up to five years as compensation for patent term lost during the FDA regulatory review process. A patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, only one patent may be extended and only those claims covering the approved drug, a method for using it, or a method for manufacturing it may be extended. Similar extensions as compensation for patent term lost during regulatory review processes are also available in certain foreign countries and territories, such as in Europe under a Supplementary Patent Certificate. However, we may not be granted an extension in the U.S. and/or foreign countries and territories because of, for example, failing to exercise due diligence during the testing phase or regulatory review process, failing to apply within applicable deadlines, failing to apply prior to expiration of relevant patents, or otherwise failing to satisfy applicable requirements. Moreover, the applicable time period or the scope of patent protection afforded could be less than we request. If we are unable to obtain a patent term extension or the term of any such extension is shorter than what we request, our competitors may obtain approval of competing products following our patent expiration, and our business, financial condition, results of operations, and growth prospects could be materially harmed.
We may be subject to claims challenging the inventorship of our patents and other intellectual property.
We may be subject to claims that former employees, scientific collaborators or other third parties have an interest in our patents, trade secrets, or other intellectual property as an inventor or co-inventor. For example, we may have inventorship disputes arise from conflicting obligations of employees, consultants, or others who are involved in developing our product candidates or other technologies. Litigation may be necessary to defend against these and other claims challenging inventorship or ownership of our patents, trade secrets, or other intellectual property. If the defense of any such claims fails, in addition to paying monetary damages, we may lose valuable intellectual property rights, such as exclusive ownership of, or right to use, intellectual property that is important to our product candidates and other technologies. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management and other employees. Any of the foregoing could have a material adverse effect on our business, financial condition, results of operations, and growth prospects.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed.
In addition to seeking patents for our product candidates and other technologies, we also rely on trade secrets and confidentiality agreements to protect our unpatented know-how, technology, and other proprietary information and to maintain
44
our competitive position. We consider trade secrets and know-how to be one of our primary sources of intellectual property. Trade secrets and know-how can be difficult to protect. We expect our trade secrets and know-how to over time be disseminated within the industry through independent development, the publication of journal articles describing the methodology, and the movement of personnel from academic to industry scientific positions.
We seek to protect these trade secrets and other proprietary technology, in part, by entering into non-disclosure and confidentiality agreements with parties who have access to them, such as our employees, corporate collaborators, outside scientific collaborators, CROs, CDMOs, consultants, advisors, and other third parties. We also enter into confidentiality and invention or patent assignment agreements with our employees and consultants as well as train our employees not to bring or use proprietary information or technology from former employers to us or in their work, and remind former employees when they leave their employment of their confidentiality obligations. We cannot guarantee that we have entered into such agreements with each party that may have or have had access to our trade secrets or proprietary technology and processes. Despite our efforts, any of these parties may breach the agreements and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive, and time-consuming, and the outcome is unpredictable. In addition, some courts inside and outside the U.S. are less willing or unwilling to protect trade secrets. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor or other third party, we would have no right to prevent them from using that technology or information to compete with us. If any of our trade secrets were to be disclosed to or independently developed by a competitor or other third party, our competitive position would be materially and adversely harmed.
We may not be successful in obtaining necessary rights to our product candidates or other technologies.
Many pharmaceutical companies, biotechnology companies, and academic institutions that compete with us in the field of neurodegeneration therapy may have patents filed and are likely filing patent applications potentially relevant to our business. In order to avoid infringing these third-party patents, we may find it necessary or prudent to obtain licenses to such patents from such third-party intellectual property holders. We may also require licenses from third parties for certain technologies for use with future product candidates. In addition, with respect to any patents we co-own with third parties, we may wish to obtain licenses to such co-owner’s interest to such patents. However, we may be unable to secure such licenses or otherwise acquire any compositions, methods of use, processes, or other intellectual property rights from third parties that we identify as necessary for our future product candidates. The licensing or acquisition of third-party intellectual property rights is a competitive area, and several more established companies may pursue strategies to license or acquire third-party intellectual property rights that we may consider attractive or necessary. These established companies may have a competitive advantage over us due to their size, capital resources, and greater clinical development and commercialization capabilities. In addition, companies that perceive us to be a competitor may be unwilling to assign or license rights to us. We also may be unable to license or acquire third-party intellectual property rights on terms that would allow us to make an appropriate return on our investment or at all. If we are unable to successfully obtain rights to required third party intellectual property rights or maintain the existing intellectual property rights we have, we may have to abandon development of the relevant program or product candidate, which could have a material adverse effect on our business, financial condition, results of operations, and growth prospects.
We may be subject to claims that our employees, consultants, or advisors have wrongfully used or disclosed alleged trade secrets of their current or former employers or claims asserting ownership of what we regard as our own intellectual property.
Many of our employees, consultants, and advisors are currently or were previously employed at universities or other biotechnology or pharmaceutical companies, including our competitors and potential competitors. Although we try to ensure that our employees, consultants, and advisors do not use the proprietary information or know-how of others in their work for us, we may be subject to claims that we or these individuals have used or disclosed intellectual property, including trade secrets or other proprietary information, of any such individual’s current or former employer. Litigation may be necessary to defend against these claims. If we fail in defending any such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. Even if we are successful in defending against such claims, litigation could result in substantial costs and be a distraction to management.
In addition, while it is our policy to require our employees and contractors who may be involved in the conception or development of intellectual property to execute agreements assigning such intellectual property to us, we may be unsuccessful
45
in executing such an agreement with each party who, in fact, conceives or develops intellectual property that we regard as our own. The assignment of intellectual property rights may not be self-executing, or the assignment agreements may be breached, and we may be forced to bring claims against third parties, or defend claims that they may bring against us, to determine the ownership of what we regard as our intellectual property. Such claims could have a material adverse effect on our business, financial condition, results of operations, and growth prospects.
Third-party claims of intellectual property infringement, misappropriation, or other violation against us may prevent or delay the development and commercialization of our product candidates and other technologies.
The field of developing innovations for neurodegenerative diseases is highly competitive and dynamic. Due to the focused research and development that is taking place by several companies, including us and our competitors, in this field, the intellectual property landscape is in flux, and it may remain uncertain in the future. Additionally, the technology used in our product candidates is still in an early stage and no products utilizing similar technology have yet reached the market. As such, there may be significant intellectual property related litigation and proceedings relating to our, and other third party, intellectual property and proprietary rights in the future.
Our commercial success depends in part on our ability to develop, manufacture, market, and sell any product candidates that we develop and to use our proprietary technologies without infringing, misappropriating, and otherwise violating the patents and other intellectual property rights of third parties. There is a substantial amount of complex litigation involving patents and other intellectual property rights in the biotechnology and pharmaceutical industries, as well as administrative proceedings for challenging patents, including interference, derivation, and reexamination proceedings before the USPTO or oppositions and other comparable proceedings in foreign jurisdictions. We may become party to, or threatened with, such actions in the future, regardless of their merit. As discussed above, recently, due to changes in U.S. law referred to as patent reform, new procedures including inter partes review and post-grant review have been implemented. As stated above, this reform adds uncertainty to the possibility of challenge to our patents in the future.
Numerous U.S. and foreign issued patents and pending patent applications owned by third parties exist in the fields in which we are developing our product candidates. As the biotechnology and pharmaceutical industries expand and more patents are issued, the risk increases that our product candidates and other technologies may give rise to claims of infringement of the patent rights of others. Although we believe that we do not infringe on any third parties’ patents or other intellectual property, we cannot assure you that our product candidates and other technologies that we have developed, are developing or may develop in the future will not infringe existing or future patents owned by third parties. We may not be aware of patents that have already been issued to a third party, such as a competitor in the fields in which we are developing product candidates, who might assert infringement of patents it may hold by our current or future product candidates or other technologies, including claims to compositions, formulations, methods of manufacture or methods of use or treatment that cover our product candidates or other technologies. It is also possible that patents owned by third parties of which we are aware, but which we do not believe are relevant to our product candidates or other technologies, could be found to be infringed by our product candidates or other technologies. In addition, because patent applications can take many years to issue, there may be currently pending patent applications that may later result in issued patents that our product candidates or other technologies may infringe.
Third parties may have patents or obtain patents in the future and claim that the manufacture, use or sale of our product candidates or other technologies infringes upon these patents. In the event that any third-party claims that we infringe their patents or that we are otherwise employing their proprietary technology without authorization and initiates litigation against us, even if we believe such claims are without merit, a court of competent jurisdiction could hold that such patents are valid, enforceable and infringed by our product candidates or other technologies. In this case, the holders of such patents may be able to block our ability to commercialize the applicable product candidate or technology unless we obtain a license under the applicable patents, or until such patents expire or are finally determined to be held invalid or unenforceable. Such a license may not be available on commercially reasonable terms or at all. Even if we are able to obtain a license, the license would likely obligate us to pay license fees or royalties or both, and the rights granted to us might be nonexclusive, which could result in our competitors gaining access to the same intellectual property. If we are unable to obtain a necessary license to a third-party patent on commercially reasonable terms, we may be unable to commercialize our product candidates or other technologies, or such commercialization efforts may be significantly delayed, which could in turn significantly harm our business.
46
Defense of infringement claims, regardless of their merit, would involve substantial litigation expense and would be a substantial diversion of management and other employee resources from our business, and may impact our reputation. In the event of a successful claim of infringement against us, we may be enjoined from further developing or commercializing our infringing product candidates or other technologies. In addition, we may have to pay substantial damages, including treble damages and attorneys’ fees for willful infringement, obtain one or more licenses from third parties, pay royalties, and/or redesign our infringing product candidates or technologies, which may be impossible or require substantial time and monetary expenditure. In that event, we would be unable to further develop and commercialize our product candidates or other technologies, which could harm our business significantly.
Engaging in litigation to defend against third parties alleging that we have infringed, misappropriated, or otherwise violated their patents or other intellectual property rights is very expensive, particularly for a company of our size, and time-consuming. Some of our competitors may be able to sustain the costs of litigation or administrative proceedings more effectively than we can because of greater financial resources. Patent litigation and other proceedings may also absorb significant management time. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings against us could impair our ability to compete in the marketplace. The occurrence of any of the foregoing could have a material adverse effect on our business, financial condition, results of operations, and growth prospects.
We may become involved in lawsuits to protect or enforce our patents and other intellectual property rights, which could be expensive, time consuming, and unsuccessful.
Competitors may infringe on our patents or the patents of our licensing partners, or we may be required to defend against claims of infringement. In addition, our patents or the patents of our licensing partners also may become involved in inventorship, priority, or validity disputes. To counter or defend against such claims can be expensive and time consuming. In an infringement proceeding, a court may decide that a patent in which we have an interest is invalid or unenforceable, the other party’s use of our patented technology falls under the safe harbor to patent infringement, or may refuse to stop the other party from using the technology at issue on the grounds that our patents do not cover the technology in question. An adverse result in any litigation proceeding could put one or more of our patents at risk of being invalidated or interpreted narrowly. Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation.
Even if resolved in our favor, litigation or other legal proceedings relating to intellectual property claims may cause us to incur significant expenses and could distract our personnel from their normal responsibilities. In addition, there could be public announcements of the results of hearings, motions, or other interim proceedings or developments, and if securities analysts or investors perceive these results to be negative, it could have a substantial adverse effect on the price of our common stock. Such litigation or proceedings could substantially increase our operating losses and reduce the resources available for development activities or any future sales, marketing, or distribution activities. We may not have sufficient financial or other resources to conduct such litigation or proceedings adequately. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their greater financial resources and more mature and developed intellectual property portfolios. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace.
Intellectual property rights do not necessarily address all potential threats.
The degree of future protection afforded by our intellectual property rights is uncertain because intellectual property rights have limitations and may not adequately protect our business or permit us to maintain our competitive advantage. For example:
|
· |
others may be able to make products that are similar to our product candidates or utilize similar technology but that are not covered by the claims of the patents that we may own; |
|
· |
we might not have been the first to make the inventions covered by the issued patent or pending patent application that we own now or in the future; |
|
· |
we might not have been the first to file patent applications covering certain of our inventions; |
47
|
· |
others may independently develop similar or alternative technologies or duplicate any of our technologies without infringing our owned intellectual property rights; |
|
· |
it is possible that our current or future pending patent applications will not lead to issued patents; |
|
· |
issued patents that we hold rights to may be held invalid or unenforceable, including as a result of legal challenges by our competitors or other third parties; |
|
· |
our competitors or other third parties might conduct research and development activities in countries where we do not have patent rights and then use the information learned from such activities to develop competitive products for sale in our major commercial markets; |
|
· |
we may not develop additional proprietary technologies that are patentable; |
|
· |
the patents of others may harm our business; and |
|
· |
we may choose not to file a patent in order to maintain certain trade secrets or know-how, and a third party may subsequently file a patent covering such intellectual property. |
Should any of these events occur, they could have a material adverse effect on our business, financial condition, results of operations, and growth prospects.
Risks Related to Our Operations
The worldwide outbreak of an infectious disease, called COVID-19, may materially and adversely affect our business operations and our ability to conduct clinical studies.
A recent, widespread outbreak of a novel infectious disease called Coronavirus Disease 2019, or COVID-19, has been declared by the World Health Organization to be a “public health emergency of international concern”; a national emergency by the President of the U.S; and a major disaster by several states in which we operate. This unprecedented spread of disease may affect our operations by causing a period of business disruption, including the potential interruption or halt of our clinical study activities and delays or disruptions in the supply of our products and product candidates, or the inability of our employees to continue their normal course of work due to disease, quarantine or leave requirements, or the possibility of legal claims and actions against us for claims of loss arising out of COVID-19. As a small company that operates with a limited number of employees, the impact of disease may disproportionately hurt our operations. Further, our business insurance may not provide coverage against economic loss or claims specifically tied to COVID-19 or any other disease.
COVID-19 presents many challenges that are without precedent. As such, we cannot presently assess or predict all current and potential uncertainties around the scope and severity of COVID-19 on our business operations with any meaningful precision. There is no assurance that COVID-19 will not have a material adverse impact on our future results. For example, the continued spread of COVID-19 could adversely impact our clinical study operations, including our ability to recruit and retain patients and principal investigators and site staff who, as healthcare providers, may have heightened exposure to COVID-19 if an outbreak occurs in their geography. COVID-19 could also negatively affect our manufacturing operations, which could result in delays or disruptions in the supply of our product candidates. A greater number of our employees working remotely during this outbreak of disease may expose us to greater risks related to cybersecurity and cyber-liability. Our study participants, vendors, employees, suppliers or others may allege they became sick due to our negligence. In addition, there could be a potential effect of a slowdown at FDA, which could result in delays of regulatory correspondence that are necessary for us to maintain or advance our product candidates in clinical studies. Further, the COVID-19 outbreak may adversely impact our ability to file on a routine and timely basis our obligations under federal securities laws, present new data at annual scientific meetings and attend professional conferences, reach out to institutional investors through in-person meetings, advance PTI-125 in a Phase 3 efficacy program, add an international component to our clinical studies, obtain additional financing as needed, engage in partnering discussions or conduct other activities necessary to the success of our business.
48
If we or any of the third parties with whom we engage were to experience shutdowns or other business disruptions related to COVID-19, our ability to conduct our business in the manner and on the timelines presently planned could be materially and negatively impacted, which could have a material adverse effect on our business, results of operations and financial condition.
We are a small company with a limited number of employees. We are highly dependent on our key personnel, and if we are not successful in attracting, motivating, and retaining highly qualified personnel, we may not be able to successfully implement our business strategy.
Our ability to compete in the highly competitive biotechnology and pharmaceutical industries depends upon our ability to attract, motivate, and retain highly qualified managerial, scientific, and medical personnel. We are highly dependent on our management, particularly our Chief Executive Officer, Remi Barbier, and our scientific and technical personnel. The loss of the services provided by any of our executive officers, other key employees, and other scientific and medical advisors, and our inability to find suitable replacements, could result in delays in the development of our product candidates and harm our business.
Competition for skilled personnel is intense and the turnover rate can be high, which may limit our ability to hire and retain highly qualified personnel on acceptable terms or at all. We expect that we may need to recruit talent from outside of our region in Austin, TX, and doing so may be costly and difficult.
To induce valuable employees to remain at our company, in addition to salary and cash incentives, we have provided equity option grants that vest over time. The value to employees of these equity grants that vest over time may be significantly affected by movements in our stock price that are beyond our control and may at any time be insufficient to counteract more lucrative offers from other companies. Although we have employment agreements with our key employees, these employment agreements provide for at-will employment, which means that any of our employees could leave our employment at any time, with or without notice. If we are unable to attract and incentivize quality personnel on acceptable terms, or at all, it may cause our business and operating results to suffer.
If our current research collaborators or scientific advisors terminate their relationships with us or develop relationships with a competitor, our ability to continue our business operations could be adversely affected.
We have relationships with research collaborators at academic and other institutions who conduct research at our request. These research collaborators are not our employees. As a result, we have limited control over their activities and, except as otherwise required by our collaboration agreements, can expect only limited amounts of their time to be dedicated to our activities. Our ability to discover drugs and biomarkers involved in human disease and validate and commercialize diagnostic tests will depend in part on the continuation of these collaborations. If any of these collaborations are terminated, we may not be able to enter into other acceptable collaborations. In addition, our existing collaborations may not be successful. Our research collaborators and scientific advisors may have relationships with other commercial entities, some of which could compete with us. Our research collaborators and scientific advisors sign agreements which provide for the confidentiality of our proprietary information and the results of studies conducted at our request. We may not, however, be able to maintain the confidentiality of our technology and other confidential information related to all collaborations. The dissemination of our confidential information could have a material adverse effect on our business.
We will need to grow the size and capabilities of our organization, and we may experience difficulties in effectively managing this growth.
As our development plans and strategies develop, we may need to add a significant number of additional managerial, operational, financial, and other personnel. Future growth will impose significant added responsibilities on members of management, including:
|
· |
identifying, recruiting, integrating, retaining, and motivating additional employees; |
|
· |
managing our internal development efforts effectively, including the clinical and FDA review process for our current and future product candidates, while complying with our contractual obligations to contractors and other third parties; |
49
|
· |
expanding our operational, financial and management controls, reporting systems, and procedures; and |
|
· |
managing increasing operational and managerial complexity. |
Our future financial performance and our ability to continue to develop and, if approved, commercialize our product candidates will depend, in part, on our ability to effectively manage any future growth. Our management may also have to divert a disproportionate amount of its attention away from day-to-day activities in order to manage these growth activities.
We currently rely, and for the foreseeable future will continue to rely, in substantial part on certain independent organizations, advisors, and consultants to provide certain services. There can be no assurance that the services of these independent organizations, advisors, and consultants will continue to be available to us on a timely basis when needed, or that we can find qualified replacements. In addition, if we are unable to effectively manage our outsourced activities or if the quality or accuracy of the services provided by consultants is compromised for any reason, our clinical studies may be extended, delayed, or terminated, and we may not be able to obtain regulatory approval of our product candidates or otherwise advance our business. There can be no assurance that we will be able to manage our existing consultants or find other competent outside contractors and consultants on economically reasonable terms, if at all.
If we are not able to effectively expand our organization by hiring new employees and expanding our groups of consultants and contractors, we may not be able to successfully implement the tasks necessary to further develop our product candidates and, accordingly, may not achieve our research, development, and commercialization goals.
Our internal computer systems, or those used by our third-party research institution collaborators, Clinical Research Organizations (CROs) or other contractors or consultants, may fail or suffer other breakdowns, cyberattacks, or information security breaches that could compromise the confidentiality, integrity, and availability of such systems and data, result in material disruptions of our development programs and business operations, risk disclosure of confidential, financial, or proprietary information, and affect our reputation.
In the ordinary course of our business, we collect and store sensitive data, including legally protected patient health information, personally identifiable information about our employees, intellectual property, and proprietary business information. We manage and maintain our applications and data utilizing on-site systems. These applications and data encompass a wide variety of business-critical information including research and development information, commercial information and business and financial information. Despite the implementation of security measures, our internal computer systems and those of our current or future CROs and other contractors and consultants may be vulnerable to damage from computer viruses and unauthorized access. As the cyber-threat landscape evolves, these attacks are growing in frequency, sophistication, and intensity, and are becoming increasingly difficult to detect. Such attacks could include the use of key loggers or other harmful and virulent malware, including ransomware or other denials of service, and can be deployed through malicious websites, the use of social engineering, and/or other means. If a breakdown, cyberattack, or other information security breach were to occur and cause interruptions in our operations, it could result in a misappropriation of confidential information, including our intellectual property or financial information, and a material disruption of our development programs and our business operations. For example, the loss of clinical study data from completed, ongoing, or future clinical studies could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. Likewise, we rely on our third-party research institution collaborators for research and development of our product candidates and other third parties for the manufacture of our product candidates and to conduct clinical studies, and similar events relating to their computer systems could also have a material adverse effect on our business. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data or systems, or inappropriate disclosure of confidential, financial, or proprietary information, including data related to our personnel, we could incur liability or risk disclosure of confidential, financial, or proprietary information, and the further development and commercialization of our product candidates could be delayed. There can be no assurance that we and our business counterparties will be successful in efforts to detect, prevent, or fully recover systems or data from all breakdowns, service interruptions, attacks, or breaches of systems that could adversely affect our business and operations and/or result in the loss of critical or sensitive data, which could result in financial, legal, business, or reputational harm to us.
Our business involves environmental risks that may result in liability for us.
In connection with our research and development activities, we, and our collaborators and vendors, are subject to federal, state and local laws, rules, regulations and policies governing the use, generation, manufacture, storage, air emission, effluent
50
discharge, handling and disposal of certain materials, biological specimens, chemicals and wastes. Although we believe that we comply with such applicable laws, regulations and policies in all material respects and have not been required to correct any material noncompliance, we may incur significant costs to comply with environmental and health and safety regulations in the future. Although we believe that our safety procedures for handling and disposing of controlled materials comply with the standards prescribed by state and federal regulations, accidental contamination or injury from these materials may occur. In the event of such an occurrence, we could be held liable for any damages that result and any such liability could exceed our resources.
Business disruptions and lack of appropriate levels of commercial insurance could seriously harm our future revenue and financial condition and increase our costs and expenses.
Our operations, and those of our third-party research institution collaborators, CROs, CDMOs, suppliers, and other contractors and consultants, could be subject to earthquakes, power shortages, telecommunications failures, water shortages, floods, hurricanes, typhoons, fires, extreme weather conditions, disease epidemics or pandemics, such as COVID-19, a novel coronavirus first detected in 2019 and for which no specific vaccine or treatment are available, and other natural or man-made disasters or business interruptions, for which we are partly or entirely uninsured. In addition, we rely on our third-party research institution collaborators for conducting research and development of our product candidates, and they may be affected by government shutdowns or withdrawn funding. The occurrence of any of these business disruptions could seriously harm our operations and financial condition and increase our costs and expenses. We rely on third party manufacturers to produce and process our product candidates. Our ability to obtain clinical supplies of our product candidates could be disrupted if the operations of these suppliers are affected by a man-made or natural disaster or other business interruption.
Our day-to-day operations are located in a single office facility in Austin, TX. Damage or extended periods of interruption to our corporate, development, or research facilities could cause us to cease or delay development of some or all our product candidates. Our insurance might not cover losses under such circumstances and our business may be seriously harmed by such delays and interruption.
Social media platforms present risks and challenges.
As social media continues to expand, it also presents us with new challenges. The inappropriate or unauthorized use of our confidential information on media platforms could cause brand damage or information leakage, which would cause legal or regulatory problems for us. In addition, negative, inappropriate or inaccurate posts or comments about us or our product candidates on social media internet sites could quickly and irreversible damage our reputation, brand image and goodwill. Further, the accidental or intentional disclosure of non-public sensitive information by our workforce or others through media channels could lead to information loss or could lead to legal or regulatory problems for us.
Risks Related to the Ownership of Our Common Stock
We do not know whether a market will continue to develop for our common stock or what the market price of our common stock will be, and, as a result, it may be difficult to sell shares of our common stock.
If a market for our common stock is not sustained, it may be difficult to sell shares of our common stock at an attractive price or at all. We cannot predict the prices at which our common stock will trade. It is possible that in one or more future periods our results of operations and progression of our product pipeline may not meet the expectations of public market analysts and investors, and, as a result of these and other factors, the price of our common stock may fall.
The market price of our common stock may be volatile, which could result in substantial losses for investors who purchase our shares.
Some of the factors that may cause the market price of our common stock to fluctuate include:
|
· |
the success of existing or new competitive products or technologies; |
|
· |
the timing and results of clinical studies for our current product candidates and any future product candidates that we may develop; |
51
|
· |
failure or discontinuation of any of our product development and research programs; |
|
· |
results of preclinical studies, clinical studies, or regulatory approvals of product candidates of our competitors, or announcements about new research programs or product candidates of our competitors; |
|
· |
regulatory or legal developments in the United States and other countries; |
|
· |
developments or disputes concerning patent applications, issued patents, or other proprietary rights; |
|
· |
the recruitment or departure of key personnel; |
|
· |
the level of expenses related to any of our research programs, clinical development programs, or product candidates that we may develop; |
|
· |
the results of our efforts to develop additional product candidates or products; |
|
· |
actual or anticipated changes in estimates as to financial results, development timelines, or recommendations by securities analysts; |
|
· |
announcement or expectation of additional financing efforts; |
|
· |
sales of our common stock by us, our insiders, or other stockholders; |
|
· |
variations in our financial results or those of companies that are perceived to be similar to us; |
|
· |
changes in estimates or recommendations by securities analysts, if any, that cover our stock; |
|
· |
market conditions in the pharmaceutical and biotechnology sectors; and |
|
· |
general economic, industry, and market conditions. |
In recent years, the stock market in general, Nasdaq, and the markets for early stage companies and pharmaceutical and biotechnology companies, has experienced significant price and volume fluctuations that have often been unrelated or disproportionate to changes in the operating performance of the companies whose stock is experiencing those price and volume fluctuations. Broad market and industry factors may seriously affect the market price of our common stock, regardless of our actual operating performance. Following periods of such volatility in the market price of a company’s securities, securities class action litigation has often been brought against that company. Because of the potential volatility of our stock price, we may become the target of securities litigation in the future. Securities litigation could result in substantial costs and divert management’s attention and resources from our business.
If securities analysts do not publish research or reports about our business or if they publish negative evaluations of our stock, the price of our stock and trading volume could decline.
The trading market for our common stock is influenced in part by the research and reports that industry or financial analysts publish about us or our business. We do not have any control over the equity research analysts that provide research coverage of our common stock or the content and opinions included in their reports. We currently have limited research coverage by industry or financial analysts, and there is no assurance that such coverage will continue. If no or few analysts maintain coverage of us, the trading price of our stock could decrease. Even if we do obtain additional analyst coverage, if one or more of the analysts covering our business downgrade their evaluations of our stock or if we fail to meet their operating results estimates for us, the price of our stock could decline. If one or more of these analysts cease to cover our stock, we could lose visibility in the market for our stock, which in turn could cause our stock price and trading volume to decline.
Raising additional capital may cause dilution to our existing stockholders, restrict our operations or require us to relinquish rights to our technologies or product candidates.
52
We may seek additional capital through a combination of public and private equity offerings, debt financings, strategic partnerships and alliances, and licensing arrangements. We, and indirectly, our stockholders, will bear the cost of issuing and servicing such securities. Because our decision to issue debt or equity securities in any future offering will depend on market conditions and other factors beyond our control, we cannot predict or estimate the amount, timing, or nature of any future offerings. To the extent that we raise additional capital through the sale of equity or debt securities, your ownership interest will be diluted, and the terms may include liquidation or other preferences that adversely affect your rights as a stockholder. The incurrence of indebtedness would result in increased fixed payment obligations and could involve restrictive covenants, such as limitations on our ability to incur additional debt, limitations on our ability to acquire, sell, or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business. Additionally, any future collaborations we enter into with third parties may provide capital in the near term but limit our potential cash flow and revenue in the future. If we raise additional funds through strategic partnerships and alliances and licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies or product candidates, or grant licenses on terms unfavorable to us.
If we are unable to maintain effective internal controls, our business, financial position, and results of operations could be adversely affected.
As a public company, we are subject to reporting and other obligations under the Securities Exchange Act of 1934, as amended (Exchange Act), including the requirements of Section 404 of SOX, which require annual management assessments of the effectiveness of our internal control over financial reporting.
The rules governing the standards that must be met for management to assess our internal control over financial reporting are complex and require significant documentation, testing and possible remediation to meet the detailed standards under the rules. During testing, our management may identify material weaknesses or deficiencies which may not be remedied in time to meet the deadline imposed by SOX. These reporting and other obligations place significant demands on our management and administrative and operational resources, including accounting resources.
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Our internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with accounting principles generally accepted in the U.S. Any failure to maintain effective internal controls could have an adverse effect on our business, financial position, and results of operations.
We are a smaller reporting company and we cannot be certain if the reduced disclosure requirements applicable to smaller reporting companies will make our common stock less attractive to investors.
We are currently a “smaller reporting company” as defined in the Exchange Act, and are thus allowed to provide simplified executive compensation disclosures in our filings and have certain other reduced disclosure obligations with respect to our SEC filings. We will remain a “smaller reporting company” until the aggregate market value of our outstanding common stock held by non-affiliates as of the last business day our recently completed second fiscal quarter is $700 million or more while our revenue remains under $100 million. We cannot predict whether investors will find our common stock less attractive because of our reliance on any of these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our stock price may be more volatile.
We cannot ensure that there will be an active, liquid trading market for our common stock, as we have in the past and may in the future fail to meet all applicable listing requirements and, our common stock may be delisted from The Nasdaq Capital Market, which could have an adverse impact on the liquidity and market price of our common stock.
Our common stock is currently listed on The Nasdaq Capital Market, which has qualitative and quantitative listing criteria that we must meet in order to remain listed on Nasdaq.
Until August 2018, we were listed on the Nasdaq Global Market and have in the past temporarily fallen out of compliance with Nasdaq listing standards and there can be no assurance that we will continue to meet the appropriate Nasdaq listing requirements in the future.
53
On March 13, 2018, we received a notice from the staff of The Nasdaq Stock Market LLC (the Staff) that we were not in compliance with Nasdaq Listing Rule 5450, setting forth the requirements for continued listing on Nasdaq. Nasdaq Listing Rule 5450 requires, among other things, that we meet one of the three standards under Nasdaq Listing Rule 5450(b); the Equity Standard; the Market Value Standard; or the Total Asset/Total Revenue Standard. The Staff notice stated that we were not in compliance with Nasdaq Listing Rule 5450(b)(2)(A) (under the Market Value Standard), as the minimum market value of our common stock had been below $50 million for 30 consecutive business days. In addition, the Staff notice stated that we did not meet the requirements under Nasdaq Listing Rule 5450(b)(3)(A) (under the Total Asset/Total Revenue Standard).
On April 26, 2018, following ten consecutive business days during which the market value of our common stock was $50 million or greater, we regained compliance with Nasdaq Listing Rule 5450(b)(2)(A).
On August 8, 2018, we received a letter from the Listing Qualifications Department of Nasdaq notifying us that our application to transfer our Nasdaq listing from the Nasdaq Global Select Market to The Nasdaq Capital Market had been approved. Our common stock was transferred to The Nasdaq Capital Market the opening of business on August 13, 2018 under the symbol “PTIE.” On March 28, 2019, our common stock symbol was changed to “SAVA”. This transfer of our listing to The Nasdaq Capital Market or any other similar future transfers could adversely affect the liquidity of our common stock. Any such event could make it more difficult to dispose of, or obtain accurate quotations for the price of, our common stock, and there also would likely be a reduction in our coverage by securities analysts, if any, and the news media, which could cause the price of our common stock to decline further. We may also face other material adverse consequences in such event, such as negative publicity, a decreased ability to obtain additional financing, diminished investor and/or employee confidence, and the loss of business development opportunities, some or all of which may contribute to a further decline in our stock price.
If future events cause our common stock to be delisted, the liquidity of our common stock would be adversely affected, investors may not be able to sell their shares quickly or at the latest market price if trading in our stock is not active, and the market price of our common stock could decrease.
Anti-takeover provisions in our charter documents and Delaware law may prevent or delay removal of incumbent management or a change of control.
Anti-takeover provisions of our amended and restated certificate of incorporation and amended and restated bylaws and Delaware law may have the effect of deterring or delaying attempts by our stockholders to remove or replace management, engage in proxy contests and effect changes in control. The provisions of our charter documents include:
|
· |
a classified board so that only one of the three classes of directors on our Board is elected each year; |
|
· |
elimination of cumulative voting in the election of directors; |
|
· |
procedures for advance notification of stockholder nominations and proposals; |
|
· |
the ability of the Board to amend our bylaws without stockholder approval; and |
|
· |
the ability of the Board to issue up to 10,000,000 shares of preferred stock without stockholder approval upon the terms and conditions and with the rights, privileges and preferences as the Board may determine. |
In addition, as a Delaware corporation, we are subject to Delaware law, including Section 203 of the Delaware General Corporation Law. In general, Section 203 prohibits a Delaware corporation from engaging in any business combination with any interested stockholder for a period of three years following the date that the stockholder became an interested stockholder unless certain specific requirements are met as set forth in Section 203.
These provisions, alone or together, could have the effect of deterring or delaying changes in incumbent management, proxy contests or changes in control.
54
Our share ownership is concentrated, and our officers, directors and principal stockholders can exert significant control over matters requiring stockholder approval.
Due to their combined stock holdings, our officers, directors and principal stockholders (i.e., stockholders holding greater than 5% of our common stock) acting collectively may have the ability to exercise significant influence over matters requiring stockholder approval including the election of directors and approval of significant corporate transactions. In particular, Remi Barbier, our founder, Chairman of the Board, President and Chief Executive Officer, owns or controls a significant amount of the voting power of our outstanding capital stock. This concentration of ownership may delay or prevent a change in control of the Company and may make some transactions, including but not limited to any merger, consolidation, or sale of substantially all of our assets, more difficult or impossible to complete without the support of key stockholders.
Publicly available information regarding stockholders’ ownership may not be comprehensive because the SEC does not require certain large stockholders to publicly disclose their stock ownership positions.
If the fair value of our stock increases and outstanding performance awards vest, we expect to use substantial amounts of cash to fund employee tax liabilities.
We have performance awards outstanding. If these performance awards vest, we expect to issue our employees shares of our common stock net of statutory employment taxes. This net issuance results in fewer shares issued and uses our cash to fund such taxes. The use of cash could be substantially higher, depending on the fair value of our common stock on the date the performance awards vest. If our use of cash to fund these taxes is substantial, our cash balance could substantially decline and our stock price could also decline.
We may in the future seek to fund the cash used for performance awards through the sale of our common stock. However, we may not be successful in selling shares of our common stock to fund the cash used for performance awards. If the number of shares we sell to fund the cash used for performance awards is significant, our stock price could decline.
Our operating results may fluctuate from quarter to quarter and this fluctuation may cause our stock price to decline.
Our quarterly operating results have fluctuated in the past and are likely to fluctuate in the future. Factors contributing to these fluctuations include, among other items, the timing and enrollment rates of clinical studies for our product candidates, our need for clinical supplies, the valuation of stock-based compensation and grant reimbursement received from NIH. Thus, quarter-to-quarter comparisons of our operating results may not be indicative of what to expect in the future. As a result, in some future quarters our clinical, financial or operating results may not meet the expectations of securities analysts and investors and could result in a decline in the price of our stock.
We may sell additional equity or debt securities to fund our operations, and have outstanding securities exercisable for our common stock, which may result in dilution to our stockholders and impose restrictions on our business.
In order to raise additional capital to support our operations, we may sell additional shares of our common stock or other securities convertible into or exchangeable for our common stock which could result in dilution our stockholders.
We cannot assure you that we will be able to sell shares or other securities in any other offering at a price per share that is equal to or greater than the price per share paid by investors in prior offerings, and investors purchasing our shares or other securities in the future could have rights superior to existing shareholders. The price per share at which we sell additional shares of our common stock or securities convertible into or exchangeable for our common stock in future transactions may be higher or lower than the price per share in prior offerings. You may also be diluted upon the exercise of warrants outstanding as of February 29, 2020 to purchase 1,615,999 shares of our common stock at an exercise price of $1.25 per share, outstanding stock options as of February 29, 2020 to purchase 3,210,965 shares of our common stock at a weighted average price of $12.27 per share, and the future issuance of up to 277,500 compensatory equity awards authorized under our 2018 Omnibus Incentive Plan and up to 58,017 shares we may sell under our Employee Stock Purchase Plan.
Changes in our ownership could limit our ability to utilize net operating loss carryforwards.
As of December 31, 2019, we had aggregate federal net operating loss carryforwards of approximately $81.5 million, which begin to expire in 2029. Under Section 382 of the Internal Revenue Code of 1986, as amended, changes in our
55
ownership may limit the amount of our net operating loss carryforwards that could be utilized annually to offset our future taxable income, if any. This limitation would generally apply in the event of a cumulative change in ownership of our company of more than 50% within a rolling three-year period. Any such limitation may significantly reduce our ability to utilize our net operating loss carryforwards and tax credit carryforwards. Any such limitation, whether as the result of past offerings, sales of our common stock by our existing stockholders or additional sales of our common stock by us in the future could have a material adverse effect on our results of operations in future years. We have not completed a study to assess whether an ownership change for purposes of Section 382 has occurred, or whether there have been multiple ownership changes since our inception, nor do we plan to do so due to the significant costs and complexities associated with such study.
Organizational Risks
We have broad discretion in the use of the net proceeds from any of our financing transactions and may not use them effectively.
Our management has broad discretion in the application of the net proceeds from our financing transactions, and investors will not have the opportunity to assess whether the net proceeds are being used appropriately. Our management could spend the net proceeds from offerings in ways that may vary substantially from their intended use, do not improve our results of operations or enhance the value of our common stock. The failure by our management to apply these funds effectively could result in financial losses that could have a material adverse effect on our business, cause the price of our common stock to decline and delay the development of our product candidates. Pending their use, we may invest the net proceeds from our financing transactions in a manner that does not produce income or that loses value.
Risks Relating to Commercialization
We currently have no in-house capabilities to manufacture or commercialize our product candidates and we rely on third-party commercial drug manufacturers for clinical drug supplies. If we are unable to develop our own manufacturing, sales, marketing and distribution capabilities, or if we are not successful in contracting with third parties for these services on favorable terms, or at all, our product revenues could be adversely impacted.
We rely on various third parties to manufacture, fill, label, store, test and ship our product candidates. We plan to continue to outsource formulation, manufacturing and related activities. These suppliers must comply with cGMP regulations enforced by the FDA and other government agencies, and are subject to ongoing periodic unannounced inspection, including preapproval inspections by the FDA and corresponding state and foreign government agencies to ensure strict compliance with cGMP and other standards. These manufacturers may subsequently be stopped from producing, manufacturing, filling, labeling, storing, testing and shipping our product candidates due to their non-compliance with federal, state or local regulations. We do not have control over our suppliers’ compliance with these regulations and standards and we cannot control decisions by our suppliers that affect their ability or willingness to continue to supply us on acceptable terms, or at all.
Disputes in the past have arisen with some of these third parties with respect to fulfilling certain conditions and obligations. There can be no guarantee that such disputes will not arise again in the future, which may lead to termination of an agreement. If an agreement is terminated, we would not be able to commercialize our product candidates until another manufacturer is identified and we have entered into a manufacturing agreement with such manufacturer. We may not be able to replace a commercial supplier on commercially reasonable terms, or at all. Replacing any of our commercial suppliers would be expensive and time consuming. Failure by any of our suppliers to perform as expected could delay or prevent the commercialization or potential regulatory approval of our product candidates for an extended period of time, result in shortages, cost overruns or other problems and would materially harm our business.
We currently have no sales, marketing or distribution capabilities. We have not established commercial strategies regarding any of our product candidates. In order to commercialize our products, if any are approved by the FDA, we will either have to develop such capabilities internally or collaborate with third parties who can perform these services for us.
56
If we decide to commercialize any of our drugs ourselves, we may not be able to
|
· |
hire and retain the necessary experienced personnel; |
|
· |
build sales, marketing and distribution operations in a cost-effective manner which are capable of successfully launching new drugs; |
|
· |
obtain access to adequate numbers of physicians to prescribe our products; or |
|
· |
generate sufficient product revenues. |
In addition, establishing such operations on our own will take time and involve significant expense. If our commercial operations lack complementary products, we may not be able to compete in a cost-effective manner with competitors with more products to sell. If we engage third-party collaborators to perform any commercial operations, our future revenues may depend significantly upon the performance of those collaborators.
If we decide to enter into new co-promotion or other licensing arrangements with third parties, we may be unable to locate acceptable collaborators because the number of potential collaborators is limited and because of competition from others for similar alliances. Even if we are able to identify one or more acceptable new collaborators, we may not be able to enter into any collaborative arrangements on favorable terms, or at all.
In addition, due to the nature of the market for our product candidates, it may be necessary for us to license all or substantially all of our product candidates to a single collaborator, thereby eliminating our opportunity to commercialize these other products independently. If we enter into any such new collaborative arrangements, our revenues are likely to be lower than if we marketed and sold our products ourselves.
In addition, any revenues we receive would depend upon our collaborators’ efforts which may not be adequate due to lack of attention or resource commitments, management turnover, change of strategic focus, business combinations or other factors outside of our control. Depending upon the terms of our collaboration, the remedies we have against an under-performing collaborator may be limited. If we were to terminate the relationship, it may be difficult or impossible to find a replacement collaborator on acceptable terms, or at all.
If physicians and patients do not accept and use our drugs, we will not achieve sufficient product revenues and our business will suffer.
Even if the FDA approves our drugs, physicians and patients may not accept and use them. Acceptance and use of our drugs will depend on a number of factors including:
|
· |
when the drug is launched into the market and related competition; |
|
· |
approved label claims; |
|
· |
perceptions by members of the healthcare community, including physicians, about the safety and effectiveness of our drugs; |
|
· |
perceptions by physicians regarding the cost benefit of our product candidates; |
|
· |
published studies demonstrating the cost-effectiveness of our drugs relative to competing products; |
|
· |
availability of reimbursement for our products from government or healthcare payers; |
|
· |
effectiveness of marketing and distribution efforts by us and other licensees and distributors. |
Because we expect to rely on sales generated by our current lead product candidates for substantially all of our revenues for the foreseeable future, the failure of any of these drugs to find market acceptance would harm our business and could require us to seek additional financing.
57
Our ability to market and promote our product candidates will be determined and limited by FDA-approved labeling.
The commercial success of our product candidates will depend upon our ability to obtain FDA-approved labeling describing their features. Our failure to achieve FDA approval of product labeling containing such information will prevent us from advertising and promoting the key features of our product candidates in order to differentiate them from other similar products. This would make our products less competitive in the market.
Risks Related to Government Regulation
If we fail to comply with the complex federal, state, local and foreign laws and regulations that apply to our business, we could suffer severe consequences that could materially and adversely affect our operating results and financial condition.
Our operations are subject to extensive federal, state, local and foreign laws and regulations, all of which are subject to change. These laws and regulations currently include, among other things:
|
· |
The Clinical Laboratory Improvement Amendments (CLIA) of 1988, which are United States federal regulatory standards that apply to all clinical laboratory testing performed on humans in the United States, requires that laboratories obtain certification from the federal government, and state licensure laws; |
|
· |
FDA laws and regulations; |
|
· |
The Health Insurance Portability and Accountability Act (HIPAA), which imposes comprehensive federal standards with respect to the privacy and security of protected health information and requirements for the use of certain standardized electronic transactions, including penalties for violators, enforcement authority to state attorneys general and requirements for breach notification; |
|
· |
state laws regulating testing and protecting the privacy of test results, as well as state laws protecting the privacy and security of health information and personal data and mandating reporting of breaches to affected individuals and state regulators; |
|
· |
the federal anti-kickback law, or the Anti-Kickback Statute, which prohibits knowingly and willfully offering, paying, soliciting, receiving, or providing remuneration, directly or indirectly, in exchange for or to induce either the referral of an individual, or the furnishing, arranging for, or recommending of an item or service that is reimbursable, in whole or in part, by a federal health care program; |
|
· |
the federal False Claims Act (FCA), which imposes liability on any person or entity that, among other things, knowingly presents, or causes to be presented, a false or fraudulent claim for payment to the federal government; |
|
· |
the federal Civil Monetary Penalties Law, which prohibits, among other things, the offering or transfer of remuneration to a Medicare or state health care program beneficiary if the person knows or should know it is likely to influence the beneficiary’s selection of a particular provider, practitioner, or supplier of services reimbursable by Medicare or a state health care program, unless an exception applies; |
|
· |
other federal and state fraud and abuse laws, such as anti-kickback laws, prohibitions on self-referral, and false claims acts, which may extend to services reimbursable by any third-party payor, including private insurers; |
|
· |
the federal Physician Payments Sunshine Act, which requires manufacturers to track and report to the federal government certain payments and other transfers of value made to physicians and teaching hospitals and ownership or investment interests held by physicians and their immediate family members; |
58
|
· |
Section 216 of the federal Protecting Access to Medicare Act of 2014 (PAMA), which requires applicable laboratories to report private payer data in a timely and accurate manner beginning in 2017 and every three years thereafter (and in some cases annually); |
|
· |
state laws that impose reporting and other compliance-related requirements; and |
|
· |
similar foreign laws and regulations that will apply to us in foreign countries in which we may choose to operate in the future. |
Enacted and future legislation may increase the difficulty and cost for us to commercialize our product candidates and may reduce the prices we are able to obtain for our product candidates.
Legislative and regulatory changes and future changes regarding the healthcare system could prevent or delay marketing approval of our product candidates, restrict or regulate post-approval activities or affect our ability to profitably sell any product candidates for which we obtain marketing approval.
In the U.S., the Medicare Prescription Drug, Improvement, and Modernization Act of 2003 (the Medicare Modernization Act) established the Medicare Part D program and provided authority for limiting the number of drugs that will be covered in any therapeutic class thereunder. The Medicare Modernization Act, including its cost reduction initiatives, could limit the coverage and reimbursement rate that we receive for any of our approved products. Private payors may follow Medicare coverage policies and payment limitations in setting their own reimbursement rates resulting in similar limits in payments from private payors.
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively, the Affordable Care Act, among other things, imposes a significant annual fee on companies that manufacture or import branded prescription product candidates. It also contains substantial provisions intended to, among other things, broaden access to health insurance, reduce or constrain the growth of health care spending, enhance remedies against healthcare fraud and abuse, add new transparency requirements for the healthcare and health insurance industries, and impose additional health policy reforms, any of which could have a material adverse effect on our business. A significant number of provisions are not yet, or have only recently become, effective, but the Affordable Care Act may result in downward pressure on pharmaceutical pricing, especially under the Medicare program, and may also increase our regulatory burdens and operating costs.
The Affordable Care Act, as well as other healthcare reform measures that have been and may be adopted in the future, may result in more rigorous coverage criteria and in additional downward pressure on the price that we receive for any approved product, and could seriously harm our future revenues. Any reduction in reimbursement from Medicare or other government programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may compromise our ability to generate revenue, attain profitability or commercialize our products.
The Affordable Care Act is a highly complex piece of legislation that continues to evolve. We do not and cannot understand or anticipate the full impact and potential implications of the Affordable Care Act on our business or on our drugs.
59
Our relationships with customers and payors will be subject to applicable anti-kickback, fraud and abuse, transparency, and other healthcare laws and regulations, which could expose us to criminal sanctions, civil penalties, exclusion from government healthcare programs, contractual damages, reputational harm, administrative burdens, and diminished profits and future earnings.
Healthcare providers, physicians and payors play a primary role in the recommendation and prescription of any product candidates for which we may obtain marketing approval. Our future arrangements with payors and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain the business or financial arrangements and relationships through which we market, sell and distribute any product candidates for which we may obtain marketing approval. Even though we do not and will not control referrals of healthcare services or bill directly to Medicare, Medicaid or other third-party payors, federal and state healthcare laws and regulations pertaining to fraud and abuse and patients' rights are and will be applicable to our business. Restrictions under applicable federal, state and foreign healthcare laws and regulations may affect our ability to operate and expose us to areas of risk, including:
|
· |
the federal Anti-Kickback Statute, which prohibits, among other things, knowingly and willfully soliciting, offering, receiving or providing remuneration, directly or indirectly, in cash or in kind, to induce or reward either the referral of an individual for, or the purchase, order or recommendation of, any good or service, for which payment may be made under federal and state healthcare programs such as Medicare and Medicaid. A person or entity does not need to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation; |
|
· |
the FCA, which imposes criminal and civil penalties, including through civil whistleblower or qui tam actions, against individuals or entities for knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government. In addition, the government may assert that a claim including items and services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the FCA; |
|
· |
HIPAA, which imposes criminal and civil liability for executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters. Similar to the federal Anti-Kickback Statute, a person or entity does not need to have actual knowledge of the statute to defraud any healthcare benefit program or specific intent to violate it in order to have committed a violation; |
|
· |
HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009 and its implementing regulations, which also imposes obligations on certain covered entity healthcare providers, health plans, and healthcare clearinghouses as well as their business associates that perform certain services involving the use or disclosure of individually identifiable health information, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information; |
|
· |
federal laws requiring drug manufacturers to report information related to payments and other transfers of value made to physicians and other healthcare providers, as well as ownership or investment interests held by physicians and their immediate family members, including under the federal Open Payments program, commonly known as the Sunshine Act, as well as other state and foreign laws regulating marketing activities; and |
|
· |
state and foreign equivalents of each of the above laws, including state anti-kickback and false claims laws, which may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non-governmental payors, including private insurers; state laws which require pharmaceutical companies to comply with the pharmaceutical industry's voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government or otherwise restricting payments that may be made to healthcare providers; and state and foreign laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts. |
60
Efforts to ensure that our business arrangements with third parties will comply with applicable healthcare laws and regulations will involve substantial costs. Nonetheless, it is possible that governmental authorities will conclude that our business practices may not comply with current or future statutes, regulations or case law involving applicable fraud and abuse or other healthcare laws and regulations. If our operations are found to be in violation of any of these laws or any other governmental regulations that may apply to us, we may be subject to significant civil, criminal and administrative penalties, damages, fines, imprisonment, exclusion from participation in government funded healthcare programs, such as Medicare and Medicaid, and the curtailment or restructuring of our operations.
Government agencies may establish and promulgate usage guidelines that could limit the use of our product candidates.
Government agencies, professional and medical societies, and other groups may establish usage guidelines that apply to our product candidates. These guidelines could address such matters as usage and dose, among other factors. Application of such guidelines could limit the clinical use or commercial appeal of our product candidates.
Risks Relating to Manufacturing
We do not own any manufacturing facilities and we rely on third-party commercial drug manufacturers for clinical drug supply.
We do not own any manufacturing facilities. We plan to continue to outsource formulation, manufacturing and related activities. We rely on a limited number of third-party suppliers to formulate, manufacture, fill, label, ship or store all of our product candidates. These suppliers must comply with current cGMP regulations enforced by the FDA and other government agencies, and are subject to ongoing periodic unannounced inspection, including preapproval inspections by the FDA and corresponding state and foreign government agencies to ensure strict compliance with cGMP and other government regulations and corresponding foreign standards. These manufacturers may subsequently be stopped from producing, storing, shipping or testing our drug products due to their non-compliance with federal, state or local regulations. We do not have control over our suppliers’ compliance with these regulations and standards. We cannot control decisions by our suppliers that affect their ability or willingness to continue to supply us on acceptable terms, or at all. We may not be able to replace a commercial supplier on commercially reasonable terms, or at all. Replacing any of our commercial suppliers would be expensive and time consuming. Failure by any of our suppliers to perform as expected could delay or prevent commercialization of our product candidates or result in shortages, cost overruns, or other problems and would materially harm our business.
Item 1B. Unresolved Staff Comments
None.
We lease approximately 6,000 square feet of office space pursuant to a non-cancelable operating lease in Austin, TX that expires December 31, 2020. We believe that our facilities are adequate and suitable for our current needs.
From time to time, we may become involved in litigation or other legal proceedings. We are not currently a party to any litigation or legal proceedings that, in the opinion of our management, are likely to have a material adverse effect on our business. Regardless of outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources, and other factors.
Item 4. Mine Safety Disclosures
Not applicable.
61
PART II
Item 5. Market for Registrant's Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market Price of Dividends of the Registrants Common Equity and Related Stockholder Matters
Our common stock is quoted on Nasdaq, under the symbol "SAVA."
We currently expect to retain future earnings, if any, for use in the operation and expansion of our business and, notwithstanding our special non-dividend distributions in December 2012 (of $0.75 per share of common stock totaling $34.0 million) and December 2010 (of $2.00 per share of common stock totaling $85.7 million), we have not paid and do not anticipate paying any cash dividends in the foreseeable future. As of January 21, 2020, there were approximately 30 registered holders of record of our common stock.
Item 6. Selected Financial Data
Not applicable.
62
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
This discussion and analysis should be read in conjunction with our financial statements and accompanying notes included elsewhere in this report. Operating results are not necessarily indicative of results that may occur in future periods.
Overview
Cassava Sciences, Inc. is a clinical stage biotechnology company. Our mission is to detect and treat neurodegenerative diseases, such as Alzheimer’s disease. Our novel science is based on stabilizing – but not removing – a critical protein in the brain.
Over the past 10 years, we have combined state-of-the-art technology with new insights in neurobiology to develop novel solutions for Alzheimer’s disease and other neurodegenerative diseases. Our strategy is to leverage our unique scientific/clinical platform to develop a first-in-class program for treating neurodegenerative diseases, such as Alzheimer’s.
We currently have two clinical-stage biopharmaceutical assets under development:
|
· |
our lead therapeutic product candidate, called PTI-125, for the treatment of Alzheimer’s disease; and |
|
· |
our lead investigational diagnostic product candidate, called SavaDx, to detect Alzheimer’s disease from a small sample of blood, possibly years before the overt appearance of clinical symptoms. |
Our scientific approach for the treatment of Alzheimer’s disease seeks to simultaneously improve both neurodegeneration and neuroinflammation. We believe our ability to improve multiple vital functions in the brain represents a new, different and crucial approach to address Alzheimer’s disease.
Our lead therapeutic product candidate, PTI-125, is a proprietary small molecule (oral) drug. PTI-125 targets an altered form of a protein called filamin A (FLNA) in the Alzheimer’s brain. Published studies have demonstrated that the altered form of FLNA causes neuronal dysfunction, neuronal degeneration and neuroinflammation.
We believe PTI-125 improves brain health by reverting altered FLNA back to its native, healthy confirmation, thus countering the downstream toxic effects of altered FLNA. We have generated and published experimental and clinical evidence of improved brain health with PTI-125. Importantly, PTI-125 is not dependent on clearing amyloid from the brain. Since PTI-125 has a unique mechanism of action, we believe its potential therapeutic effects may be additive or synergistic with that of other therapeutic candidates aiming to treat neurodegeneration.
PTI-125 has demonstrated a multitude of beneficial effects in animal models of disease, including normalizing neurotransmission, decreasing neuroinflammation, suppressing neurodegeneration, and restoring memory and cognition.
In 2019, we completed a small, first-in-human, clinical-proof-of-concept, open-label Phase 2a study of PTI-125 in the U.S., with substantial support from the National Institute on Aging (NIA), a division of the NIH. Treatment with PTI-125 for 28 days significantly improved key biomarkers of Alzheimer’s pathology, neurodegeneration and neuroinflammation (p<0.001). Biomarkers effects were seen in all patients in both CSF and plasma. To our knowledge, no other drug candidate has improved an entire panel of biomarkers of in patients with Alzheimer’s disease.
A confirmatory, randomized, placebo-controlled, multi-center Phase 2b study of PTI-125 in Alzheimer’s disease is on-going as of March 2020. We expect to announce Phase 2b results in approximately mid-2020.
Our diagnostic effort, called SavaDx, is an early-stage program focused on detecting Alzheimer’s disease from a small sample of blood, possibly years before the overt appearance of clinical symptoms. We are developing SavaDx as a fast, accurate and quantitative blood-based investigational biomarker/diagnostic to detect and monitor Alzheimer's disease. The goal is to make the detection of Alzheimer’s disease as simple as getting a blood test.
Alzheimer’s disease is a progressive neurodegenerative disorder that affects cognition, function and behavior. An estimated 5.8 million Americans are living with Alzheimer’s disease in 2020, according to the Alzheimer’s Association, a non-profit organization. There are no disease-modifying drug therapies to treat the disease.
63
PTI-125 and SavaDx were both discovered and designed in-house and were characterized by our academic collaborators during research activities that were conducted from approximately 2008 to date. We own exclusive, worldwide rights to these drug assets and related technologies, without royalty obligations to any third party. Our patent protection in this area currently runs through 2034.
Financial Overview
We have yet to generate any revenues from product sales. We have an accumulated deficit of $168.6 million at December 31, 2019. These losses have resulted principally from costs incurred in connection with research and development activities, salaries and other personnel-related costs and general corporate expenses. Research and development activities include costs of preclinical and clinical studies as well as clinical supplies associated with our product candidates. Salaries and other personnel-related costs include non-cash stock-based compensation associated with options and other equity awards granted to employees and non-employees. Our operating results may fluctuate substantially from period to period as a result of the timing of preclinical activities, enrollment rates of clinical studies for our product candidates and our need for clinical supplies.
We believe that our cash and cash equivalents at December 31, 2019, will enable us to fund our operating expenses for at least the next 12 months. In addition, we may seek in the future to fund our operations through additional public or private equity or debt financings or other sources. However, we may be unable to raise additional funds or enter into such other arrangements when needed on favorable terms or at all. If we are unable to obtain financing or reach profitability, the related lack of liquidity will have a material adverse effect on our operations and future prospects.
We expect to continue to use significant cash resources in our operations for the next several years. Our cash requirements for operating activities and capital expenditures may increase substantially in the future as we:
|
· |
conduct preclinical and clinical studies for our product candidates; |
|
· |
seek regulatory approvals for our product candidates; |
|
· |
develop, formulate, manufacture and commercialize our product candidates; |
|
· |
implement additional internal systems and develop new infrastructure; |
|
· |
acquire or in-license additional products or technologies, or expand the use of our technology; |
|
· |
maintain, defend and expand the scope of our intellectual property; and |
|
· |
hire additional personnel. |
Product revenue will depend on our ability to receive regulatory approvals for, and successfully market, our product candidates. If our development efforts result in regulatory approval and successful commercialization of our product candidates, we will generate revenue from direct sales of our drugs and/or, if we license our drugs to future collaborators, from the receipt of license fees and royalties from sales of licensed products. We conduct our research and development programs through a combination of internal and collaborative programs. We rely on arrangements with universities, our collaborators, CROs and clinical research sites for a significant portion of our product development efforts.
64
The following table summarizes expenses which have been reduced for reimbursements received for NIH grants (in thousands):
|
|
||||||
|
|
Twelve months ended |
|||||
|
|
December 31, |
|||||
|
|
2019 |
2018 |
||||
|
Research and development expenses - gross |
$ |
6,297 |
$ |
6,016 | ||
|
Less: Reimbursement from NIH grants |
4,729 | 3,047 | ||||
|
Research and development expenses - net |
$ |
1,568 |
$ |
2,969 | ||
|
|
||||||
Research and development expenses include compensation, contractor fees and supplies as well as allocated common costs. Contractor fees and supplies generally include expenses for preclinical studies and clinical studies and costs for formulation and manufacturing activities. Other common costs include the allocation of common costs such as facilities. During the year ended December 31, 2019 and 2018, we received $4.7 million and $3.0 million from research grants from the NIH. These reimbursements were recorded as a reduction to our research and development expenses.
Our technology has been applied across certain of our portfolio of product candidates. Data, know-how, personnel, clinical results, research results and other matters related to the research and development of any one of our product candidates also relate to, and further the development of, our other product candidates. As a result, costs allocated to a specific product candidate may not necessarily reflect the actual costs surrounding research and development of such product candidate due to cross application of the foregoing.
Estimating the dates of completion of clinical development, and the costs to complete development, of our product candidates would be highly speculative, subjective and potentially misleading. Pharmaceutical products take a significant amount of time to research, develop and commercialize. The clinical study portion of the development of a new drug alone usually spans several years. We expect to reassess our future research and development plans based on our review of data we receive from our current research and development activities. The cost and pace of our future research and development activities are linked and subject to change.
Critical Accounting Policies
The preparation of our financial statements in accordance with U.S. generally accepted accounting principles requires us to make estimates and assumptions that affect the reported amounts of assets, liabilities, revenues, expenses and interest income in our financial statements and accompanying notes. We evaluate our estimates on an ongoing basis, including those estimates related to agreements and research collaborations. We base our estimates on historical experience and various other assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results
65
may differ from these estimates under different assumptions or conditions. The following items in our financial statements require significant estimates and judgments:
|
· |
Stock-based compensation. We recognize non-cash expense for the fair value of all stock options and other share-based awards. We use the Black-Scholes option valuation model to calculate the fair value of stock options, using the single-option award approach and straight-line attribution method. The Company adopted ASU No. 2018-07, Compensation—Stock Compensation (Topic 718), Improvements to Nonemployee Share-Based Payment Accounting, on January 1, 2019. Accordingly, for all options granted, it recognizes the resulting fair value as expense on a straight-line basis over the vesting period of each respective stock option, generally four years. |
We have granted share-based awards that vest upon achievement of certain performance criteria, or performance awards. We multiply the number of performance awards by the fair market value of our common stock on the date of grant to calculate the fair value of each award. We estimate an implicit service period for achieving performance criteria for each award. We recognize the resulting fair value as expense over the implicit service period when we conclude that achieving the performance criteria is probable. We periodically review and update as appropriate our estimates of implicit service periods and determinations on achievement of the performance criteria. Performance awards vest and common stock is issued upon achievement of the performance criteria.
|
· |
Income Taxes. The Company accounts for income taxes under the asset and liability method. Deferred tax assets and liabilities are recognized for the estimated future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases. Deferred tax balances are adjusted to reflect tax rates based on currently enacted tax laws, which will be in effect in the years in which the temporary differences are expected to reverse. The Company has accumulated significant deferred tax assets that reflect the tax effects of net operating loss and tax credit carryovers and temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Realization of certain deferred tax assets is dependent upon future earnings. The Company is uncertain about the timing and amount of any future earnings. Accordingly, the Company offsets these deferred tax assets with a valuation allowance. |
The Company accounts for uncertain tax positions in accordance with ASC 740, “Income Taxes”, which clarifies the accounting for uncertainty in tax positions. These provisions require recognition of the impact of a tax position in the Company’s financial statements only if that position is more likely than not of being sustained upon examination by taxing authorities, based on the technical merits of the position. Any interest and penalties related to uncertain tax positions will be reflected as a component of income tax expense.
|
· |
Research Contract Costs and Accruals. The Company has entered into various research and development contracts with research institutions and other third-party vendors. These agreements are generally cancelable, and related payments are recorded as research and development expenses as incurred. The Company records accruals for estimated ongoing research costs. When evaluating the adequacy of the accrued liabilities, the Company analyzes progress of the studies including the phase or completion of events, invoices received and contracted costs. Significant judgments and estimates are made in determining the accrued balances at the end of any reporting period. Actual results could differ from the Company’s estimates. The Company’s historical accrual estimates have not been materially different from the actual costs. |
Recent Accounting Pronouncements
In December 2019, the FABS issued ASU No. 2019-12, Income Taxes (Topic 740) Simplifying Accounting for Income Taxes as part of its initiative to reduce complexity in the accounting standards. The guidance amended certain disclosure requirements that had become redundant, outdated or superseded. Additionally, this guidance amends accounting for the interim period effects of changes in tax laws or rates, and simplifies aspects of the accounting for franchise taxes. The guidance is effective for annual periods beginning after December 15, 2020, and is applicable for the Company in fiscal 2021. Early adoption is permitted. The Company does not anticipate that this guidance will have a material impact on its consolidated financial statements.
66
In November 2018, the FASB issued ASU No. 2018-18, Collaborative Arrangements (Topic 808): Clarifying the Interaction Between Topic 808 and Topic 606. The amendments in this update provide guidance on how to assess whether certain transactions between collaborative arrangement participants should be accounted for within the revenue recognition standard. The amendments in this update are effective for interim and annual periods for the Company beginning on January 1, 2020. The Company does not anticipate that this guidance will have a material impact on its consolidated financial statements.
In August 2018, the FASB issued ASU No. 2018-13, Fair Value Measurement (Topic 820) - Disclosure Framework - Changes to the Disclosure Requirements for Fair Value Measurement (ASU 2018-13), which is designed to improve the effectiveness of disclosures by removing, modifying and adding disclosures related to fair value measurements. ASU 2018-13 is effective for fiscal years beginning after December 15, 2019, including interim periods within those fiscal years. The Company is currently evaluating the impact of ASU 2018-13 on its consolidated financial statements.
Results of Operations
Research and Development Expense
Research and development expense consist primarily of costs of drug development work associated with our product candidates, including:
|
· |
preclinical testing, |
|
· |
clinical studies, |
|
· |
clinical supplies and related formulation and design costs, and |
|
· |
compensation and other personnel-related expenses. |
Research and development expenses decreased to $1.6 million in 2019 from $3.0 million in 2018, representing a 47% decrease. This decrease was attributable to reimbursements of $4.7 million received from research grants in 2019 from the NIH and recorded as a reduction to research and development expense, as compared to $3.0 million in recorded reimbursements in 2018, and decreased non-cash stock-related compensation expenses of $0.5 million in 2019 compared to $1.0 million in 2018, offset by $0.8 million in increased research and development expenses compared to the prior year due to costs incurred for our Phase 2 clinical programs.
We expect research and development expense to fluctuate in future periods as we continue our development efforts. We expect our development efforts to result in our product candidates progressing through various stages of clinical studies. Our research and development expenses may fluctuate from period to period due to the timing and scope of our development activities and the results of clinical studies and preclinical studies.
General and Administrative Expense
General and administrative expenses consist of personnel costs, allocated expenses and other expenses for outside professional services, including legal, human resources, audit and accounting services. Personnel costs consist of salaries, bonus, benefits and stock-based compensation. Allocated expenses consist primarily of facility costs. We incur expenses associated with operating as a public company, including expenses related to compliance with the rules and regulations of the SEC and Nasdaq, additional insurance expenses, additional audit expenses, investor relations activities, SOX compliance expenses and other administrative expenses and professional services. General and administrative expense decreased to $3.4 million in 2019 from $3.7 million in 2018, representing an 8% decrease. This was due primarily to decreases in non-cash stock-based compensation expenses offsetting an increase in compensation costs from the hiring of a chief financial officer in October 2018.
General and administrative expenses included non-cash stock-based compensation expenses of $0.8 million in 2019 compared to $1.4 million in 2018. We expect other general and administrative expense for 2020 will be consistent with 2019.
67
Interest Income
Interest and other income, net, was $328,000 in 2019 compared to $105,000 in 2018. The increase was due primarily to higher cash balances from our August 2018 stock offering as well as fluctuations in interest rates.
Liquidity and Capital Resources
Since inception, we have financed our operations primarily through public and private stock offerings, payments received under collaborative agreements and interest earned on our investments. We intend to continue to use our capital resources to fund research and development activities, capital expenditures, working capital requirements and other general corporate purposes. As of December 31, 2019, cash and cash equivalents totaled $23.1 million.
2018 Registered Direct Offering
On August 17, 2018, we completed a common stock offering pursuant to which certain investors purchased 8.9 million shares of common stock and warrants at a price of $1.275 per share. Net proceeds of the offering were approximately $10.1 million after deducting offering expenses. The warrants are exercisable for 8.9 million shares of common stock at $1.25 per share. Subject to certain ownership limitations described in the warrants, the warrants were immediately exercisable and will remain exercisable until February 17, 2021. The warrants will be exercisable on a “cashless” basis in certain circumstances, including while there is no effective registration statement registering the shares of common stock issuable upon exercise of the warrants at any time until the expiry of the warrants. Such registration statement was declared effective by the SEC on January 30, 2019. The warrants provide that holders will have the right to participate in any rights offering or distribution of assets together with the holders of common stock on an as-exercised basis.
In conjunction with the offering, we also issued to the placement agent warrants to purchase up to 0.3 million shares of common stock (the Placement Agent Warrants). The Placement Agent Warrants have substantially the same terms as the warrants to be issued to the purchasers of common stock, except that their exercise price is $1.59 per share.
During 2019, we received proceeds of $5.9 million from the exercise of 4.6 million warrants. Subsequently, from December 31, 2019 to March 26, 2020, we received proceeds of $3.6 million from the exercise of an additional 2.9 million warrants. As of March 26, 2020, 1.6 million warrants remain outstanding, each with a strike price of $1.25 per share. All warrants outstanding expire February 17, 2021.
At the Market (ATM) Common Stock Issuance
On February 8, 2018, we established an at-the-market offering program (ATM) to sell, from time to time, shares of our common stock in transactions pursuant to a shelf registration statement that was declared effective by the SEC on July 31, 2017. During the year ended December 31, 2018, we sold a total of 1,763,013 shares of our common stock under the ATM Agreement in the open market for net proceeds of $3.9 million.
We sold no shares under the ATM in 2019. We terminated the ATM on February 20, 2020.
NIH Research Grant Awards
Our research has been supported by NIH under multiple research grant awards. Strong, long-term support from NIH has allowed us to advance our two lead product candidates, PTI-125 and SavaDx, into clinical development.
During the year ended December 31, 2018, we were awarded two NIH grants totaling up to $6.7 million to support Phase II programs with PTI-125.
In March 2020, we were awarded a supplemental research funding grant from NIH of up to $374,000. This new, non-dilutive research funding is intended to strengthen our clinical program of PTI-125 in patients with Alzheimer’s disease.
Use of Cash
68
Net cash used in operating activities was $2.5 million for 2019, resulting primarily from a $4.6 million net loss incurred partially offset by $1.3 million of stock-based compensation expense and $0.8 million from changes in operating assets and liabilities.
Net cash used in operating activities was $4.8 million for 2018, resulting primarily from a $6.6 million net loss incurred partially offset by $2.4 million of stock-based compensation expense. Net cash used in operating activities also included $0.7 million of cash used from changes in operating assets and liabilities.
Cash used in investing activities was $18,000 for 2019 from purchases of computer equipment. There was no cash from investing activities in 2018.
Net cash provided by financing activities was $5.8 million consisting of $5.9 million proceeds from exercise of common stock warrants partially offset by $0.1 million in offering expenses related the 2018 sale of common stock and warrants.
Net cash provided by financing activities was $14.1 million in 2018 consisting of net proceeds from the sale of common stock and warrants in August 2018 as well as the sale of common stock under our At-The-Market facility, as described above.
Realization of our deferred tax assets is dependent on future earnings, if any. We are uncertain about the timing and amount of any future earnings. Accordingly, we offset these net deferred tax assets with a valuation allowance.
We lease approximately 6,000 square feet of office space pursuant to a non-cancelable operating lease in Austin, TX that expires in 2020. Future minimum lease payments are $0.1 million at December 31, 2019.
We have an accumulated deficit of $168.6 million at December 31, 2019. We expect our cash requirements to be significant in the future. The amount and timing of our future cash requirements will depend on regulatory and market acceptance of our product candidates and the resources we devote to researching and developing, formulating, manufacturing, commercializing and supporting our products. We believe that our current resources should be sufficient to fund our operations for at least the next 12 months. We may seek additional future funding through public or private financing in the future, if such funding is available and on terms acceptable to us.
Off-balance Sheet Arrangements
As of December 31, 2019, we did not have any relationships with unconsolidated entities or financial partnerships, such as entities often referred to as structured finance or special purpose entities, which would have been established for the purpose of facilitating off-balance sheet arrangements or other contractually narrow or limited purposes. In addition, we do not engage in trading activities involving non-exchange traded contracts. Therefore, we are not materially exposed to financing, liquidity, market or credit risk that could arise if we had engaged in these relationships. We do not have relationships or transactions with persons or entities that derive benefits from their non-independent relationship with us or our related parties.
Item 7A. Quantitative and Qualitative Disclosures about Market Risk
Pursuant to Item 305(e) of Regulation S-K, the information called for by Item 7A is not required.
69
Item 8. Financial Statements and Supplementary Data
INDEX TO FINANCIAL STATEMENTS
|
|
Page |
|||
|
71 |
||||
|
72 |
||||
|
73 |
||||
|
|
|
|||
|
74 |
||||
|
75 |
||||
|
76 |
||||
70
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of Cassava Sciences, Inc.
Opinion on the Financial Statements
We have audited the accompanying balance sheets of Cassava Sciences, Inc. (the Company) as of December 31, 2019 and 2018, the related statements of operations, stockholders' equity and cash flows for the years then ended, and the related notes (collectively referred to as the “financial statements”). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company at December 31, 2019 and 2018, and the results of its operations and its cash flows for the years then ended, in conformity with U.S. generally accepted accounting principles.
Basis for Opinion
These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits we are required to obtain an understanding of internal control over financial reporting but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
/s/ Ernst & Young LLP
We have served as the Company’s auditor since 2002.
Austin, Texas
March 26, 2020
71
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CASSAVA SCIENCES, INC. |
|||||
|
|
|
|
|
|
|
|
BALANCE SHEETS |
|||||
|
(In thousands, except share and par value data) |
|||||
|
|
|
|
|
|
|
|
|
December 31, |
||||
|
|
2019 |
|
2018 |
||
|
ASSETS |
|||||
|
Current assets: |
|
|
|
|
|
|
Cash and cash equivalents |
$ |
23,081 |
|
$ |
19,807 |
|
Other current assets |
|
268 |
|
|
233 |
|
Total current assets |
|
23,349 |
|
|
20,040 |
|
Operating lease right-of-use assets |
|
90 |
|
|
— |
|
Property and equipment, net |
|
47 |
|
|
87 |
|
Other assets |
|
— |
|
|
12 |
|
Total assets |
$ |
23,486 |
|
$ |
20,139 |
|
|
|
|
|
|
|
|
LIABILITIES AND STOCKHOLDERS' EQUITY |
|||||
|
Current liabilities: |
|
|
|
|
|
|
Accounts payable |
$ |
453 |
|
$ |
294 |
|
Accrued development expense |
|
777 |
|
|
156 |
|
Accrued compensation and benefits |
|
58 |
|
|
61 |
|
Operating lease liabilities, current |
|
90 |
|
|
— |
|
Other current liabilities |
|
9 |
|
|
— |
|
Total current liabilities |
|
1,387 |
|
|
511 |
|
Operating lease liabilities, non-current |
|
— |
|
|
— |
|
Total liabilities |
|
1,387 |
|
|
511 |
|
Commitments and contingencies |
|
|
|
|
|
|
Stockholders' equity: |
|
|
|
|
|
|
Preferred stock, $.001 par value; 10,000,000 shares authorized, none issued and outstanding |
|
— |
|
|
— |
|
Common stock, $.001 par value; 120,000,000 shares authorized; 21,841,810 and 17,219,300 shares issued and outstanding at December 31, 2019 and 2018, respectively |
|
22 |
|
|
17 |
|
Additional paid-in capital |
|
190,664 |
|
|
183,567 |
|
Accumulated deficit |
|
(168,587) |
|
|
(163,956) |
|
Total stockholders' equity |
|
22,099 |
|
|
19,628 |
|
Total liabilities and stockholders' equity |
$ |
23,486 |
|
$ |
20,139 |
|
|
|
|
|
|
|
See accompanying notes to financial statements.
72
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CASSAVA SCIENCES, INC. |
||||||
|
|
|
|
|
|
|
|
|
STATEMENTS OF OPERATIONS |
||||||
|
(In thousands, except per share data) |
||||||
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
|
Year ended December 31, |
||||
|
|
|
2019 |
|
2018 |
||
|
Operating expenses: |
|
|
|
|
|
|
|
Research and development, net of grant reimbursement |
|
$ |
1,568 |
|
$ |
2,969 |
|
General and administrative |
|
|
3,391 |
|
|
3,693 |
|
Total operating expenses |
|
|
4,959 |
|
|
6,662 |
|
Operating loss |
|
|
(4,959) |
|
|
(6,662) |
|
Interest income |
|
|
328 |
|
|
105 |
|
Net loss |
|
$ |
(4,631) |
|
$ |
(6,557) |
|
Net loss per share, basic and diluted |
|
$ |
(0.27) |
|
$ |
(0.61) |
|
Shares used in computing net loss per share, basic and diluted |
|
|
17,412 |
|
|
10,682 |
|
|
|
|
|
|
|
|
See accompanying notes to financial statements.
73
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
CASSAVA SCIENCES, INC. |
||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
STATEMENTS OF STOCKHOLDERS' EQUITY |
||||||||||||||||
|
(in thousands) |
||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Accumulated |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
other |
|
|
|
|
Total |
|||||
|
|
Common stock |
|
Additional |
|
comprehensive |
|
Accumulated |
|
stockholders' |
|||||||
|
|
Shares |
|
Par value |
|
paid-in capital |
|
income |
|
deficit |
|
equity |
|||||
|
Balance at December 31, 2017 |
6,595,509 |
|
$ |
7 |
|
$ |
167,091 |
|
$ |
— |
|
$ |
(157,399) |
|
$ |
9,699 |
|
Non-cash stock-related compensation for: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stock options for employees |
— |
|
|
— |
|
|
2,352 |
|
|
— |
|
|
— |
|
|
2,352 |
|
Stock options for non-employees |
— |
|
|
— |
|
|
36 |
|
|
— |
|
|
— |
|
|
36 |
|
Issuance of common stock and warrants |
10,623,791 |
|
|
10 |
|
|
14,088 |
|
|
— |
|
|
— |
|
|
14,098 |
|
Net loss |
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(6,557) |
|
|
(6,557) |
|
Balance at December 31, 2018 |
17,219,300 |
|
$ |
17 |
|
$ |
183,567 |
|
$ |
— |
|
$ |
(163,956) |
|
$ |
19,628 |
|
Non-cash stock-related compensation for: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stock options for employees |
— |
|
|
— |
|
|
1,287 |
|
|
— |
|
|
— |
|
|
1,287 |
|
Stock options for non-employees |
— |
|
|
— |
|
|
9 |
|
|
— |
|
|
— |
|
|
9 |
|
Issuance costs from 2018 sale of common stock and warrants |
— |
|
|
— |
|
|
(60) |
|
|
— |
|
|
— |
|
|
(60) |
|
Issuance of common stock pursuant to exercise of warrants |
4,622,510 |
|
|
5 |
|
|
5,861 |
|
|
— |
|
|
— |
|
|
5,866 |
|
Net loss |
— |
|
|
— |
|
|
— |
|
|
— |
|
|
(4,631) |
|
|
(4,631) |
|
Balance at December 31, 2019 |
21,841,810 |
|
$ |
22 |
|
$ |
190,664 |
|
$ |
— |
|
$ |
(168,587) |
|
$ |
22,099 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
See accompanying notes to financial statements.
74
|
|
|||||
|
CASSAVA SCIENCES, INC. |
|||||
|
|
|
|
|
|
|
|
STATEMENTS OF CASH FLOWS |
|||||
|
(In thousands) |
|||||
|
|
|
|
|
|
|
|
|
|
||||
|
|
Year ended December 31, |
||||
|
|
2019 |
|
2018 |
||
|
Cash flows from operating activities: |
|
|
|
|
|
|
Net loss |
$ |
(4,631) |
|
$ |
(6,557) |
|
Adjustments to reconcile net loss to net cash used in operating activities: |
|
|
|
|
|
|
Non-cash stock-based compensation |
|
1,296 |
|
|
2,388 |
|
Depreciation and amortization |
|
58 |
|
|
69 |
|
Changes in operating assets and liabilities: |
|
|
|
|
|
|
Other current assets |
|
(23) |
|
|
(49) |
|
Accounts payable |
|
159 |
|
|
(130) |
|
Accrued development expense |
|
621 |
|
|
(243) |
|
Accrued compensation and benefits |
|
(3) |
|
|
(248) |
|
Other current liabilities |
|
9 |
|
|
— |
|
Net cash used in operating activities |
|
(2,514) |
|
|
(4,770) |
|
Cash flows from investing activities: |
|
|
|
|
|
|
Purchases of property and equipment |
|
(18) |
|
|
— |
|
Net cash used in investing activities |
|
(18) |
|
|
— |
|
Cash flows from financing activities: |
|
|
|
|
|
|
Issuance costs from 2018 sale of common stock and warrants |
|
(60) |
|
|
— |
|
Proceeds from sale of common stock and warrants, net |
|
— |
|
|
14,098 |
|
Proceeds from exercise of common stock warrants, net |
|
5,866 |
|
|
— |
|
Net cash provided by financing activities |
|
5,806 |
|
|
14,098 |
|
Net increase in cash and cash equivalents |
|
3,274 |
|
|
9,328 |
|
Cash and cash equivalents at beginning of period |
|
19,807 |
|
|
10,479 |
|
Cash and cash equivalents at end of period |
$ |
23,081 |
|
$ |
19,807 |
|
|
|||||
See accompanying notes to financial statements.
75
NOTES TO FINANCIAL STATEMENTS
1. General and Liquidity
Cassava Sciences, Inc. (the Company), formerly known as Pain Therapeutics, Inc., develops proprietary drugs that offer significant improvements to patients and healthcare professionals. The Company generally focuses its drug development efforts on disorders of the nervous system.
Liquidity
The Company has incurred significant net losses and negative cash flows since inception, and as a result has an accumulated deficit of $168.6 million at December 31, 2019. The Company expects its cash requirements to be significant in the future. The amount and timing of its future cash requirements will depend on regulatory and market acceptance of the Company’s product candidates and the resources it devotes to researching and developing, formulating, manufacturing, commercializing and supporting its products. The Company may seek additional future funding through public or private financing in the future, if such funding is available and on terms acceptable to the Company.
2. Summary of Significant Accounting Policies
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires that management make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amount of revenue earned and expenses incurred during the reporting period. Actual results could differ from those estimates.
Proceeds from Grants
In 2019, the Company received $4.7 million of reimbursement from the National Institutes of Health and National Institute on Drug Abuse and $3.0 million in 2018. The Company records the proceeds from these grants as reductions to its research and development expenses.
Cash and Cash Equivalents and Concentration of Credit Risk
The Company invests in cash and cash equivalents and, in the past, marketable securities. The Company considers highly-liquid financial instruments with original maturities of three months or less to be cash equivalents. Highly liquid investments that are considered cash equivalents include money market funds, certificates of deposit, treasury bills and commercial paper. The carrying value of cash equivalents approximates fair value due to the short-term maturity of these securities.
The Company’s investment policy allows for investments in marketable securities with active secondary or resale markets, establishes diversification and credit quality requirements and limits investments by maturity and issuer. The Company maintains its investments at one financial institution.
76
Fair Value Measurements
The Company reports its cash and cash equivalents at fair value as Level 1, Level 2 or Level 3 using the following inputs:
|
· |
Level 1 includes quoted prices in active markets. The Company bases the fair value of money market funds and U.S. treasury securities on Level 1 inputs. |
|
· |
Level 2 includes significant observable inputs, such as quoted prices for identical or similar investments, or other inputs that are observable and can be corroborated by observable market data for similar securities. The Company uses market pricing and other observable market inputs obtained from third-party providers. It uses the bid price to establish fair value where a bid price is available. The Company bases the fair value of its certificates of deposit and marketable securities, if any, on Level 2 inputs. |
|
· |
Level 3 includes unobservable inputs that are supported by little or no market activity. The Company does not have any investments where the fair value is based on Level 3 inputs. |
If a financial instrument uses inputs that fall in different levels of the hierarchy, the instrument will be categorized based upon the lowest level of input that is significant to the fair value calculation. Certificates of deposit are included within cash equivalents as a Level 2 input. The fair value of the remainder of cash and cash equivalents was based on Level 1 inputs at December 31, 2019 and 2018.
Business Segments
The Company reports segment information based on how it internally evaluates the operating performance of its business units, or segments. The Company’s operations are confined to one business segment: the development of novel drugs and diagnostics.
Stock-based Compensation
The Company recognizes non-cash expense for the fair value of all stock options and other share-based awards. The Company uses the Black-Scholes option valuation model to calculate the fair value of stock options, using the single-option award approach and straight-line attribution method. This model requires the input of subjective assumptions including expected stock price volatility, expected life and estimated forfeitures of each award. These assumptions consist of estimates of future market conditions, which are inherently uncertain, and therefore, are subject to management's judgment.
The Company adopted ASU No. 2018-07, Compensation—Stock Compensation (Topic 718), Improvements to Nonemployee Share-Based Payment Accounting, on January 1, 2019. Accordingly, for all options granted, it recognizes the resulting fair value as expense on a straight-line basis over the vesting period of each respective stock option, generally four years.
The Company has granted share-based awards that vest upon achievement of certain performance criteria, or performance awards. The Company multiplies the number of performance awards by the fair market value of its common stock on the date of grant to calculate the fair value of each award. It estimates an implicit service period for achieving performance criteria for each award. The Company recognizes the resulting fair value as expense over the implicit service period when it concludes that achieving the performance criteria is probable. The Company periodically reviews and updates as appropriate its estimates of implicit service periods and conclusions on achieving the performance criteria. Performance awards vest and common stock is issued upon achievement of the performance criteria.
77
Net Loss per Share
Basic net loss per share is computed on the basis of the weighted-average number of common shares outstanding for the reporting period. Diluted net loss per share is computed on the basis of the weighted-average number of common shares outstanding plus dilutive potential common shares outstanding using the treasury-stock method. Potential dilutive common shares consist of outstanding equity awards and warrants. There is no difference between the Company’s net loss and comprehensive loss. The numerators and denominators in the calculation of basic and diluted net loss per share were as follows (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||
|
|
Year ended December 31, |
|
||||
|
|
2019 |
|
2018 |
|
||
|
Numerator: |
|
|
|
|
|
|
|
Net loss |
$ |
(4,631) |
|
$ |
(6,557) |
|
|
Denominator: |
|
|
|
|
|
|
|
Shares used in computing net loss per share, basic and diluted |
|
17,412 |
|
|
10,682 |
|
|
Net loss per share, basic and diluted |
$ |
(0.27) |
|
$ |
(0.61) |
|
|
|
|
|
|
|
|
|
The Company excluded weighted equity awards and warrants outstanding to purchase common stock from the calculation of diluted net loss per share because the effect of including these shares in this calculation would be anti-dilutive.
The potential shares of common stock that have been excluded from the diluted loss per share calculation above for the years ended December 31, 2019 and 2018 were as follows:
|
|
|||
|
|
Year ended December 31, |
||
|
|
2019 |
|
2018 |
|
Stock options |
2,932,421 |
|
2,864,754 |
|
Stock warrants |
4,504,091 |
|
9,126,601 |
|
Potential common shares |
7,436,512 |
|
11,991,355 |
|
|
|
|
|
Fair Value of Financial Instruments
Financial instruments include cash and cash equivalents, accounts payable and accrued liabilities. The estimated fair value of financial instruments has been determined using available market information or other appropriate valuation methodologies. However, considerable judgment is required in interpreting market data to develop estimates of fair value; therefore, the estimates are not necessarily indicative of the amounts that could be realized or would be paid in a current market exchange. The effect of using different market assumptions and/or estimation methodologies may be material to the estimated fair value amounts. The carrying amounts of cash and cash equivalents, accounts payable and accrued liabilities are at cost, which approximates fair value due to the short maturity of those instruments.
Right-of-use Asset and Liability
The Company has a single non-cancelable operating lease for approximately 6,000 square feet of office space in Austin, Texas that expires on December 31, 2020, which is used for the development of novel drugs. Prior to January 1, 2019, the Company accounted for leases in accordance with the provisions of ASC Topic 840. Under the previous leasing guidance, the Company expensed lease payments over the term of the lease and did not give recognition to any lease related assets or liabilities on its balance sheet.
On January 1, 2019, the Company adopted ASU No. 2016-02, Leases (ASC 842) which, as permitted by ASC Topic 842, is the date of initial application. The core principle of ASC Topic 842 is that a lessee should recognize the assets and liabilities that arise from leases. For operating leases, a lessee is required to recognize a right-of-use asset and a lease liability, initially measured at the present value of the lease payments, in the statement of financial position. The Company recognized a right-of-use asset and operating lease liability upon the adoption of ASU 2016-02 which increased total
78
assets and total liabilities relative to such amounts prior to adoption. The Company utilized a discount rate of 5.5% to determine the present value of the future lease payments which represents the Company’s incremental borrowing rate.
The impact of adopting ASC 842 on assets and liabilities recorded as of January 1, 2019 were as follows (in thousands):
|
|
||
|
|
|
|
|
Assets |
|
|
|
Operating lease right-of-use assets |
$ |
180 |
|
|
|
|
|
Liabilities |
|
|
|
Operating lease liabilities, current |
|
90 |
|
Operating lease liabilities, non-current |
$ |
90 |
|
|
The Company recorded a reduction of the non-current portion of the lease liability and an offsetting reduction in the right-of-use assets of $90,000 during the year ended December 31, 2019. There was no change to the statement of operations during the year ended December 31, 2019 or statement of cash flows during the year ended September 30, 2019 as a result of the adoption of ASC Topic 842. See additional information regarding leases in Note 8 – Commitments and Contingencies.
Income Taxes
The Company accounts for income taxes under the asset and liability method. Deferred tax assets and liabilities are recognized for the estimated future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases. Deferred tax balances are adjusted to reflect tax rates based on currently enacted tax laws, which will be in effect in the years in which the temporary differences are expected to reverse. The Company has accumulated significant deferred tax assets that reflect the tax effects of net operating loss and tax credit carryovers and temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Realization of certain deferred tax assets is dependent upon future earnings. The Company is uncertain about the timing and amount of any future earnings. Accordingly, the Company offsets these deferred tax assets with a valuation allowance.
The Company accounts for uncertain tax positions in accordance with ASC 740, “Income Taxes”, which clarifies the accounting for uncertainty in tax positions. These provisions require recognition of the impact of a tax position in the Company’s financial statements only if that position is more likely than not of being sustained upon examination by taxing authorities, based on the technical merits of the position. Any interest and penalties related to uncertain tax positions will be reflected as a component of income tax expense.
Research Contract Costs and Accruals
The Company has entered into various research and development contracts with research institutions and other third-party vendors. These agreements are generally cancelable, and related payments are recorded as research and development expenses as incurred. The Company records accruals for estimated ongoing research costs. When evaluating the adequacy of the accrued liabilities, the Company analyzes progress of the studies including the phase or completion of events, invoices received and contracted costs. Significant judgments and estimates are made in determining the accrued balances at the end of any reporting period. Actual results could differ from the Company’s estimates. The Company’s historical accrual estimates have not been materially different from the actual costs.
Recent Accounting Pronouncements
The Company reviewed recently issued accounting pronouncements and plan to adopt those that are applicable to it, and does not expect the adoption of these pronouncements to have a material impact on its financial position, results of operations or cash flows.
79
In December 2019, the FABS issued ASU No. 2019-12, Income Taxes (Topic 740) Simplifying Accounting for Income Taxes as part of its initiative to reduce complexity in the accounting standards. The guidance amended certain disclosure requirements that had become redundant, outdated or superseded. Additionally, this guidance amends accounting for the interim period effects of changes in tax laws or rates, and simplifies aspects of the accounting for franchise taxes. The guidance is effective for annual periods beginning after December 15, 2020, and is applicable for the Company in fiscal 2021. Early adoption is permitted. The Company does not anticipate that this guidance will have a material impact on its consolidated financial statements.
In November 2018, the FASB issued ASU No. 2018-18, Collaborative Arrangements (Topic 808): Clarifying the Interaction Between Topic 808 and Topic 606. The amendments in this update provide guidance on how to assess whether certain transactions between collaborative arrangement participants should be accounted for within the revenue recognition standard. The amendments in this update are effective for interim and annual periods for the Company beginning on January 1, 2020. The Company does not anticipate that this guidance will have a material impact on its consolidated financial statements.
In August 2018, the FASB issued ASU No. 2018-13, Fair Value Measurement (Topic 820) - Disclosure Framework - Changes to the Disclosure Requirements for Fair Value Measurement (ASU 2018-13), which is designed to improve the effectiveness of disclosures by removing, modifying and adding disclosures related to fair value measurements. ASU 2018-13 is effective for fiscal years beginning after December 15, 2019, including interim periods within those fiscal years. The Company is currently evaluating the impact of ASU 2018-13 on its consolidated financial statements.
3. Collaboration Agreement
The Company had formerly entered into a Development and License Agreement (the License Agreement) with Durect Corporation around certain controlled-release technology. On March 20, 2019, the Company gave notice of termination for such License Agreement. This and other actions effectively ended the Company’s development of any product candidates related to such technology.
4. Property and Equipment
The Company’s property and equipment include furniture and equipment with a purchase value of $1.0 million at December 31, 2019 and 2018. Depreciation is recognized using the straight-line method over the expected life of the property and equipment. Accumulated depreciation was $0.9 million at December 31, 2019 and 2018.
Subsequent to December 31, 2019, the Company completed the sale, to an independent third party, of fully depreciated surplus manufacturing equipment and received net proceeds totaling $100,000.
5. Stockholders' Equity and Stock-Based Compensation
Preferred Stock
The Company’s Board of Directors has the authority to issue preferred stock in one or more series and to fix the rights, preferences, privileges, restrictions and the number of shares constituting any series or the designation of the series.
2018 Registered Direct Offering
On August 17, 2018, the Company completed a common stock offering pursuant to which certain investors purchased 8,860,778 shares of common stock and warrants at a price of $1.275 per share. Net proceeds of the offering were approximately $10.1 million after deducting offering expenses. The warrants are exercisable for 8,860,778 shares of common stock at $1.25 per share. Subject to certain ownership limitations described in the warrants, the warrants are exercisable until February 17, 2021. The warrants will be exercisable on a “cashless” basis in certain circumstances, including while there is no effective registration statement registering the shares of common stock issuable upon exercise of the warrants until the expiry of the warrants. Such registration statement was declared effective by the SEC on January 30, 2019. The warrants provide that holders will have the right to participate in any rights offering or distribution of assets together with the holders of Common Stock on an as-exercised basis.
80
In conjunction with the offering, the Company also issued to the placement agent warrants to purchase up to 265,823 shares of common stock (the Placement Agent Warrants). The Placement Agent Warrants have substantially the same terms as the warrants to be issued to the purchasers of common stock, except that their exercise price is $1.59 per share.
During 2019, the Company received proceeds of $5.9 million from the exercise of 4.6 million shares pursuant to warrants. Subsequent to December 31, 2019 and through March 26, 2020, the Company received proceeds of $3.6 million from the exercise of an additional 2.9 million shares pursuant to warrants.
Warrants outstanding as of December 31, 2019 and 2018 were as follows:
|
|
|
|
|
|
|
Number of Shares Outstanding under Warrant |
||
|
|
|
|
|
|
|
December 31, |
||
|
Issuance Date |
|
Expiration Date |
|
Exercise Price Per Share |
|
2019 |
|
2018 |
|
|
|
|
|
|
|
|
|
|
|
August 17, 2018 |
|
February 17, 2021 |
|
1.25 |
|
4,496,116 |
|
8,860,778 |
|
August 17, 2018 |
|
February 17, 2021 |
|
1.59 |
|
7,975 |
|
265,823 |
|
|
|
|
|
|
|
4,504,091 |
|
9,126,601 |
|
|
|
|
|
|
|
|
|
|
The sale of common stock and issuance of warrants qualified for equity treatment under GAAP. The respective values of the warrants and common stock were calculated using their relative fair values and classified under common stock and additional paid-in capital. The value ascribed to the warrants is $7.2 million and to the common stock is approximately $3.0 million.
The fair value of these warrants was estimated using a Black-Scholes model with the following assumptions: estimated volatility 136%, risk-free interest rate of 2.65%, no dividend and an expected life of 2.5 years.
At the Market (ATM) Common Stock Issuance
In February 2018, the Company established an at-the-market offering program (ATM) to sell, from time to time, shares of Company common stock in transactions pursuant to a shelf registration statement that was declared effective by the SEC on July 31, 2017.
There were no sales of common stock under the ATM Agreement during the year ended December 31, 2019. During the year ended December 31, 2018, the Company sold a total of 1,763,013 shares of its common stock under the ATM Agreement in the open market for net proceeds of $3.9 million. The Company terminated the ATM on February 20, 2020.
2008 Equity Incentive Plan
Under the Company’s 2008 Equity Incentive Plan, or 2008 Equity Plan, its employees, directors and consultants received share-based awards, including grants of stock options and performance awards. The 2008 Equity Plan expired in December 2017. Share-based awards generally expire ten years from the date of grant.
2018 Equity Incentive Plan
On January 31, 2018, the Company’s Board of Directors approved the Company’s 2018 Omnibus Incentive Plan (the 2018 Plan). The Company’s Board of Directors or a designated Committee of the Board is responsible for administration of the 2018 Plan and determined the terms and conditions of each option granted, consistent with the terms of the 2018 Plan. The Company’s employees, directors, and consultants are eligible to receive awards under the 2018 Plan, including grants of stock options and performance awards. Share-based awards generally expire ten years from the date of grant. The 2018 Plan provides for issuance of up to 1,000,000 shares of common stock, par value $0.001 per share under the 2018 Plan, subject to adjustment as provided in the 2018 Plan.
When stock options or performance awards are exercised net of the exercise price and taxes, the number of shares of stock issued is reduced by the number of shares equal to the amount of taxes owed by the award recipient and that number
81
of shares are cancelled. The Company then uses its cash to pay tax authorities the amount of statutory taxes owed by and on behalf of the award recipient.
Stock Options
The following summarizes information about stock option activity during 2019:
|
|
|
|
Number of Options |
|
|
Weighted Average Exercise Price |
|
Weighted Average Remaining Contractual Term |
|
Aggregate Intrinsic Value |
|
|
|
|
|
|
|
|
|
|
In years |
|
In millions |
|
|
|
Outstanding as of December 31, 2018 |
|
2,964,973 |
|
$ |
14.14 |
|
6.06 |
|
$ |
0.0 |
|
|
Options granted |
|
425,000 |
|
|
1.80 |
|
|
|
|
|
|
|
Options exercised |
|
— |
|
|
— |
|
|
|
|
|
|
|
Options forfeited/canceled |
|
(179,008) |
|
|
18.37 |
|
|
|
|
|
|
|
Outstanding as of December 31, 2019 |
|
3,210,965 |
|
|
12.27 |
|
6.05 |
|
$ |
4,153,583 |
|
|
Vested and expected to vest at December 31, 2019 |
|
3,210,965 |
|
|
12.27 |
|
6.05 |
|
$ |
4,153,583 |
|
|
Exercisable at December 31, 2019 |
|
2,143,886 |
|
$ |
17.06 |
|
4.70 |
|
$ |
1,258,713 |
|
|
|
|
|
|
|
|
|
|
|
|
|
The following summarizes information about stock options at December 31, 2019 by a range of exercise prices:
|
|
|||||||||||||||||
|
|
|
|
|
|
|
|
Options outstanding |
|
Options exercisable |
||||||||
|
|
|
|
|
|
|
|
|
|
Weighted |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
average |
|
Weighted |
|
|
|
Weighted |
||
|
|
|
|
|
|
|
|
Number of |
|
remaining |
|
average |
|
Number of |
|
average |
||
|
|
Range of exercise prices |
|
outstanding |
|
contractual |
|
exercise |
|
vested |
|
exercise |
||||||
|
|
From |
|
To |
|
options |
|
life (in years) |
|
price |
|
options |
|
price |
||||
|
|
$ |
0.95 |
|
$ |
1.88 |
|
707,500 |
|
9.4 |
|
$ |
1.48 |
|
93,487 |
|
$ |
1.02 |
|
|
$ |
3.24 |
|
$ |
4.09 |
|
892,575 |
|
7.7 |
|
$ |
3.56 |
|
497,433 |
|
$ |
3.54 |
|
|
$ |
4.10 |
|
$ |
16.66 |
|
688,323 |
|
5.2 |
|
$ |
12.04 |
|
631,412 |
|
$ |
12.44 |
|
|
$ |
16.87 |
|
$ |
35.00 |
|
779,691 |
|
2.7 |
|
$ |
25.03 |
|
778,678 |
|
$ |
25.03 |
|
|
$ |
36.40 |
|
$ |
53.55 |
|
142,876 |
|
1.6 |
|
$ |
51.65 |
|
142,876 |
|
$ |
51.65 |
|
|
|
|
|
|
|
|
3,210,965 |
|
6.1 |
|
$ |
12.27 |
|
2,143,886 |
|
$ |
17.06 |
The Company uses Black-Scholes to estimate the fair value of options granted. Black-Scholes considers a number of factors, including the market price of the Company’s common stock. For options granted to employees and directors, it used certain factors to value each stock option granted, which resulted in a weighted average fair value of options granted during 2019 and 2018, as follows:
|
|
|
2019 |
|
2018 |
|
|
Volatility |
118% to 119% |
|
87% to 118% |
|
|
Risk-free interest rates |
1.7% to 2.5% |
|
2.7% to 2.9% |
|
|
Expected life of option |
7 years |
|
7 years |
|
|
Dividend yield |
zero |
|
zero |
|
|
Forfeiture rate |
zero |
|
zero |
|
|
Weighted average fair value of stock options granted |
$1.60 |
|
$0.98 |
|
|
Volatility is based on reviews of the historical volatility of the Company’s common stock. Risk-free interest rates are based on yields of U.S. treasury notes in effect at the date of grant. Expected life of option is based on actual historical option exercises. Dividend yield is zero because the Company does not anticipate paying cash dividends in the foreseeable future.
82
For options granted to non-employees, the Company estimates the fair value of stock options granted using factors similar to those used for stock options granted to employees and directors and appropriate for the terms underlying the stock options granted to non-employees.
As of December 31, 2019, the Company expects to recognize compensation expense of $2.1 million related to non-vested options held by employees and directors over the weighted average remaining recognition period of 2.7 years.
Performance Awards
The following summarizes information about performance award activity during 2019:
|
|
|
Number of Performance Awards |
|
Outstanding as of December 31, 2018 |
|
138,055 |
|
Granted |
|
— |
|
Vested performance awards |
|
— |
|
Forfeited/Canceled |
|
— |
|
Outstanding as of December 31, 2019 |
|
138,055 |
|
|
|
|
If and when outstanding performance awards vest, the Company would recognize $2.3 million in non-cash stock-based compensation expense. These performance awards expire between 2022 and 2026.
Stock-Based Compensation Expense
The following summarizes information about non-cash stock-based compensation expense, in thousands:
|
|
||||||
|
|
|
Twelve months ended |
||||
|
|
|
December 31, |
||||
|
|
|
2019 |
|
2018 |
||
|
Research and development |
|
$ |
542 |
|
$ |
1,043 |
|
|
|
|
|
|
|
|
|
General and administrative |
|
|
754 |
|
|
1,345 |
|
|
|
|
|
|
|
|
|
Total non-cash stock-based compensation expense |
|
$ |
1,296 |
|
$ |
2,388 |
|
|
|
|
|
|
|
|
6. Employee 401(k) Benefit Plan
The Company has a defined-contribution savings plan under Section 401(k) of the Internal Revenue Code. The plan covers substantially all employees. Employees are eligible to participate in the plan the first day of the month after hire and may contribute up to the current statutory limits under Internal Revenue Service regulations. The 401(k) plan permits the Company to make additional matching contributions on behalf of all employees. Through December 31, 2019, the Company has not made any matching contributions to the 401(k) plan.
7. Income Taxes
U.S. Tax Reform
In December 2017, legislation commonly known as the Tax Cuts and Jobs Act, or the Tax Act, was signed into law. The Tax Act included a number of changes to existing tax law, including, among other changes, a permanent reduction the U.S. federal corporate tax rate from 35% to 21%, effective as of January 1, 2018, as well as limitation of the deduction for net operating losses to 80% of annual taxable income and elimination of net operating loss carrybacks, in each case, for losses arising in taxable years beginning after December 31, 2017 (though any such net operating losses may be carried forward indefinitely).
In connection with the Tax Act, the Company remeasured certain deferred tax assets and liabilities based on the rates at which they are expected to reverse in the future, which is generally 21%. The remeasurement of the Company's deferred tax balance was primarily offset by application of its valuation allowance. The Company completed its
83
accounting for all of the enactment date income tax effects of the Tax Act as of December 31, 2018 and recognized no material adjustments.
The Company did not provide for income taxes in 2019 and 2018 because it had a net operating loss for tax purposes in those years and the tax benefit that would have resulted from the statutory rate was fully offset by the valuation allowance.
The reconciliation of the statutory federal income tax rate to the Company’s effective tax rate for the years ended December 31, 2019 and 2018 was as follows:
|
|
||||||||||
|
|
Year ended December 31, |
|||||||||
|
|
2019 |
2018 |
||||||||
|
Tax at federal statutory rate |
21.0 |
% |
21.0 |
% |
||||||
|
State tax, net of federal benefit |
- |
- |
||||||||
|
Equity-based compensation |
(2.4) | (2.9) | ||||||||
|
Research & development credits |
0.6 | 1.9 | ||||||||
|
Change in valuation allowance |
(19.2) | (20.0) | ||||||||
|
Effective income tax rate |
— |
% |
— |
% |
||||||
|
|
||||||||||
Deferred tax assets and valuation allowance
Deferred tax assets reflect the tax effects of net operating loss and tax credit carryforwards and temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. The Company’s deferred taxes assets at December 31, 2019 and 2018 were valued at the corporate tax rate of 21% to reflect the Tax Act. The Company offsets its deferred tax assets by a valuation allowance because it is uncertain about the timing and amount of any future profits. Significant components of its deferred tax assets are as follows (in thousands):
|
|
December 31, |
||||
|
|
2019 |
2018 |
|||
|
Deferred tax assets: |
|||||
|
Net operating loss carryforwards |
$ |
17,110 |
$ |
16,500 | |
|
Stock-related compensation |
5,817 | 5,700 | |||
|
Research & development credit carryforwards |
6,587 | 6,500 | |||
|
Other |
237 | 100 | |||
|
Total deferred tax assets: |
29,751 | 28,800 | |||
|
Valuation allowance |
(29,725) | (28,800) | |||
|
Net deferred tax assets: |
$ |
26 |
$ |
— |
|
|
Deferred tax liabilities: |
|||||
|
Property and equipment |
$ |
(7) |
$ |
— |
|
|
Operating lease right-of-use assets |
(19) |
— |
|||
|
Total deferred tax liabilities: |
$ |
(26) |
$ |
— |
|
|
Net deferred tax asset / (liability): |
$ |
— |
$ |
— |
|
|
|
|||||
The valuation allowance increased by $0.9 million and $1.3 million in 2019 and 2018, respectively, due to additional book losses in the periods.
The Company’s pre-tax net operating loss carryforwards of $81.5 million are federal, of which $74.1 million expires between 2029 and 2037 and $7.4 million carries forward indefinitely. As of December 31, 2019, the Company had federal research and development tax credits of approximately $11.0 million, which expire in the years 2023 through 2038.
84
Unrecognized tax benefits
The Company has unrecognized tax benefits related to tax credits. The Company added to its unrecognized tax benefits in 2019 and 2018 as follows (in thousands):
|
|
|||||
|
|
Year ended December 31, |
||||
|
|
2019 |
2018 |
|||
|
Beginning balance |
$ |
4,400 |
$ |
4,300 | |
|
Additions based on tax positions related to the current year |
— |
100 | |||
|
Ending balance |
$ |
4,400 |
$ |
4,400 | |
|
|
|||||
As of December 31, 2019, there were no unrecognized tax benefits that we expect would change significantly over the next 12 months.
The Company files U.S. and Texas income tax returns. In the United States, the statute of limitations with respect to the federal income tax returns for tax years after 2015 are open to audit; however, since the Company has net operating losses, the taxing authority has the ability to review tax returns prior to the 2016 tax year and make adjustments to these net operating loss carryforwards. We are not under audit in any taxing jurisdiction at this time.
8. Leases and Commitments
The Company leases approximately 6,000 square feet of office space pursuant to a non-cancelable operating lease in Austin, TX that expires on December 31, 2020. Future minimum lease payments are (in thousands).
|
|
|||
|
For the year ending December 31, |
|
|
|
|
2020 |
|
|
99 |
|
Total future minimum lease payments |
|
$ |
99 |
|
Lease: imputed interest |
|
|
(9) |
|
Total |
|
$ |
90 |
|
|
|
|
|
The Company believes that its facilities are adequate and suitable for its current needs. Rent expense was $0.1 million both in 2019 and 2018.
The Company conducts its product research and development programs through a combination of internal and collaborative programs that include, among others, arrangements with universities, contract research organizations and clinical research sites. It has contractual arrangements with these organizations, however these contracts are cancelable on thirty days’ notice and the Company’s obligations under these contracts are largely based on services performed.
85
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Evaluation of disclosure controls and procedures.
Our management, with the participation of our Chief Executive Officer and our Chief Financial Officer, evaluated the effectiveness of our disclosure controls and procedures as of the end of the period covered by this Annual Report on Form 10-K. Based on this evaluation, our Chief Executive Officer and our Chief Financial Officer have concluded that our disclosure controls and procedures are effective to ensure that information we are required to disclose in reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission, or SEC, rules and forms and that such information is accumulated and communicated to management as appropriate to allow timely decisions regarding required disclosures.
Management’s annual report on internal control over financial reporting. Our management is responsible for establishing and maintaining adequate internal control over our financial reporting. Our management has assessed the effectiveness of internal control over financial reporting as of December 31, 2019. Our assessment was based on criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission, or COSO, in Internal Control-Integrated Framework (2013 Framework).
Our internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Our internal control over financial reporting includes those policies and procedures that:
|
(1) |
pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect our transactions and dispositions of our assets; |
|
(2) |
provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of our management and board of directors; and |
|
(3) |
provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of our assets that could have a material effect on the financial statements. |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Based on the COSO criteria, we believe our internal control over financial reporting as of December 31, 2019 was effective.
This Annual Report on Form 10-K does not include an attestation report of our independent registered public accounting firm regarding internal control over financial reporting. Management’s assessment of the effectiveness of our internal control over financial reporting as of December 31, 2019, was not subject to attestation by our independent registered public accounting firm pursuant to rules of the SEC that permit a smaller reporting company to provide only management’s report in the Company’s Annual Report on Form 10-K.
Changes in internal control over financial reporting.
There was no change in our internal control over financial reporting that occurred during the quarter ended December 31, 2019 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
86
None.
PART III
Item 10. Directors and Executive Officers and Corporate Governance
The information regarding our directors, executive officers, director nomination process and the audit committee of the Board is incorporated by reference from "Directors and Executive Officers" in our Proxy Statement for our 2020 Annual Meeting of Stockholders.
Section 16(a) Beneficial Ownership Reporting Compliance
Section 16(a) of the Exchange Act requires our executive officers and directors and persons who own more than ten percent (10%) of a registered class of our equity securities to file reports of ownership and changes in ownership with the SEC. Executive officers, directors and greater than ten percent (10%) stockholders are required to furnish us with copies of all Section 16(a) forms they file. We believe all of our executive officers and directors complied with all applicable filing requirements during 2019.
Code of Ethics
We have adopted a Code of Ethics that applies to all of our directors, officers and employees, including our principal executive officer and principal financial officer. We publicize the Code of Ethics through posting the policy on our website, http://www.cassavasciences.com. We will disclose on our website any waivers of, or amendments to, our Code of Ethics.
Item 11. Executive Compensation
The information required by this Item is incorporated by reference from our definitive Proxy Statement referred to in Item 10 above where it appears under the heading "Executive Compensation and Other Matters."
Item 12.Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The information required by this Item regarding security ownership of certain beneficial owners and management is incorporated by reference from our definitive Proxy Statement referred to in Item 10 above where it appears under the heading "Security Ownership of Certain Beneficial Owners and Management."
The following table summarizes the securities authorized for issuance under our equity compensation plans as of December 31, 2019:
|
|
|
Number of |
|
|
Weighted Average |
|
Number of Securities |
|
|
Equity compensation plans approved by stockholders |
|
3,349,020 |
(1) |
$ |
11.77 |
(2) |
335,517 |
(3) |
|
Equity compensation plans not approved by stockholders |
|
— |
|
|
— |
|
— |
|
|
|
|
3,349,020 |
|
$ |
11.77 |
|
335,517 |
|
|
|
|
|
|
|
|
|
|
|
|
(1) |
Includes outstanding stock options and awards for 2,626,520 shares of our common stock under the 2008 Plan and 722,500 shares of our common stock under the 2018 Plan. |
|
(2) |
Includes the weighted average stock price for outstanding stock options of $15.36 under the 2008 Plan and $1.65 for the 2018 Plan. |
87
|
(3) |
Represents 277,500 shares of our common stock for the 2018 Plan and 58,017 for the Employee Stock Purchase Plan. No future awards shall occur under the 2008 Plan. |
Item 13. Certain Relationships and Related Transactions and Director Independence
The information required by this Item is incorporated by reference from our definitive Proxy Statement referred to in Item 10 above where it appears under the heading "Certain Relationships and Related Transactions."
Item 14. Principal Accountant Fees and Services
The information required by this Item is incorporated by reference from our definitive Proxy Statement referred to in Item 10 above where it appears under the heading “Principal Accountant Fees and Services.”
88
Item 15. Exhibits and Financial Statement Schedules
(a)The following documents are filed as part of this Form 10-K:
(1)Financial Statements (included in Part II of this report):
Report of Independent Registered Public Accounting Firm
Balance Sheets
Statements of Operations
Statements of Stockholders' Equity
Statements of Cash Flows
Notes to Financial Statements
(2)Financial Statement Schedules:
All financial statement schedules are omitted because the information is inapplicable or presented in the notes to the financial statements.
(3)Management Contracts, Compensatory Plans and Arrangements.
Management contracts, compensatory plans and arrangements are indicated by the symbol “*” in the applicable exhibits listed in Item 15(b), below.
(b)Exhibits
The exhibits listed below are filed as part of this Form 10-K other than Exhibit 32.1, which shall be deemed furnished.
89
|
|
|
Agreement between Registrant and H.C. Wainwright & Co., dated August 15, 2018. |
|
8-K |
|
8/20/2018 |
|
10.2 |
|
|
|
|
* |
|
Employment Agreement, executed on October 9, 2018, by and between Registrant and Eric Schoen. |
|
8-K |
|
10/11/2018 |
|
10.1 |
|
|
|
|
23.1 |
|
|
|
|
|
|
|
|
|
X |
|
|
24.1 |
|
|
Power of Attorney (included in the signature page to this report). |
|
|
|
|
|
|
|
X |
|
31.1 |
|
|
|
|
|
|
|
|
|
X |
|
|
31.2 |
|
|
|
|
|
|
|
|
|
X |
|
|
32.1 |
|
|
|
|
|
|
|
|
|
X |
|
|
101.INS |
|
|
XBRL Instance Document. |
|
|
|
|
|
|
|
X |
|
101.SCH |
|
|
XBRL Taxonomy Extension Schema Document. |
|
|
|
|
|
|
|
X |
|
101.CAL |
|
|
XBRL Taxonomy Extension Calculation Linkbase Document. |
|
|
|
|
|
|
|
X |
|
101.DEF |
|
|
XBRL Taxonomy Extension Definition Linkbase Document. |
|
|
|
|
|
|
|
X |
|
101.LAB |
|
|
XBRL Taxonomy Extension Labels Linkbase Document. |
|
|
|
|
|
|
|
X |
|
101.PRE |
|
|
XBRL Taxonomy Extension Presentation Linkbase Document. |
|
|
|
|
|
|
|
X |
|
* Management contract, compensatory plan or arrangement. |
|
|
|
|
|
|
|
|
|||
|
+ Portions of this Exhibit are subject to a confidential treatment order. |
|
|
|
|
|
|
|
|
|||
(c)Financial Statement Schedules
All financial statement schedules are omitted because the information is inapplicable or presented in the notes to the financial statements.
The Company has elected not to include summary information.
90
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
|
|
Cassava Sciences, Inc. |
|
|
|
(Registrant) |
|
|
|
|
|
|
|
/s/ REMI BARBIER |
|
|
|
Remi Barbier, |
|
|
|
Chairman of the Board of Directors, |
|
|
|
President and Chief Executive Officer |
|
|
|
|
|
Dated: March 26, 2020
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Remi Barbier his true and lawful attorneys-in-fact, with full power of substitution, for him in any and all capacities, to sign any amendments to this Annual Report on Form 10-K and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, hereby ratifying and confirming all that each of said attorneys-in-fact or their substitute or substitutes may do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
|
|
|
|
|
|
|
|
|
|
|
|
|
Signature |
|
Title |
|
Date |
|
|
|
|
|
|
|
/s/ REMI BARBIER |
|
President, Chief Executive Officer and |
|
March 26, 2020 |
|
Remi Barbier |
|
Chairman of the Board of Directors |
|
|
|
|
|
(Principal Executive Officer) |
|
|
|
|
|
|
|
|
|
/s/ ERIC J. SCHOEN |
|
Chief Financial Officer |
|
March 26, 2020 |
|
Eric J. Schoen |
|
(Principal Financial Officer and Principal Accounting Officer) |
|
|
|
|
|
|
|
|
|
/s/ NADAV FRIEDMANN, PH.D., M.D. |
|
Chief Operating and Medical Officer |
|
March 26, 2020 |
|
Nadav Friedmann, Ph.D., M.D. |
|
and Director |
|
|
|
|
|
|
|
|
|
/s/ ROBERT Z. GUSSIN, PH.D. |
|
Director |
|
March 26, 2020 |
|
Robert Z. Gussin, Ph.D. |
|
|
|
|
|
|
|
|
|
|
|
/s/ MICHAEL J. O'DONNELL, ESQ. |
|
Director |
|
March 26, 2020 |
|
Michael J. O'Donnell, Esq. |
|
|
|
|
|
|
|
|
|
|
|
/s/ SAIRA RAMASASTRY |
|
Director |
|
March 26, 2020 |
|
Saira Ramasastry |
|
|
|
|
|
|
|
|
|
|
|
/s/ SANFORD R. ROBERTSON |
|
Director |
|
March 26, 2020 |
|
Sanford R. Robertson |
|
|
|
|
|
|
|
|
|
|
|
/s/ PATRICK SCANNON, M.D., PH.D. |
|
Director |
|
March 26, 2020 |
|
Patrick Scannon, M.D., Ph.D. |
|
|
|
|
|
|
|
|
|
|
91