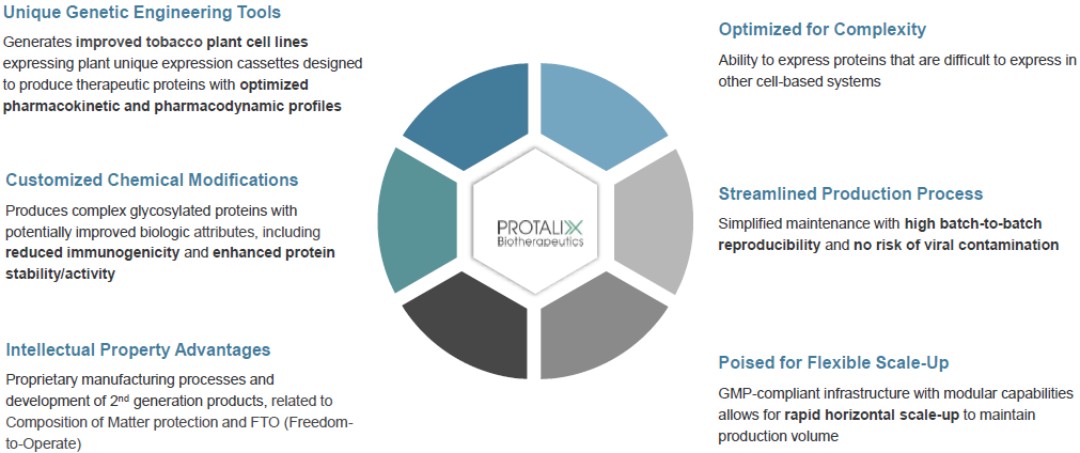
ProCellEx consists of a comprehensive set of proprietary technologies and capabilities, including the use of advanced genetic engineering and plant cell culture technology, enabling us to produce complex, proprietary, and biologically equivalent proteins for a variety of human diseases. Our protein expression system facilitates the creation and selection of high-expressing, genetically-stable cell lines capable of expressing recombinant proteins. The system plays an important role in the execution of our corporate strategy as its allows us to develop and produce tailored complex recombinant therapeutic proteins and to genetically engineer and/or chemically modify such proteins pre- and/or post-production. The engineering and modification of the therapeutic proteins have the potential to provide added clinical benefits by improving the biological characteristics (e.g., glycosylation, half-life, immunogenicity).
Our ProCellEx technology allows for many unique advantages, including: biologic optimization; an ability to handle complex protein expressions; flexible manufacturing with improvements through efficiencies, enhancements and/or rapid horizontal scale-ups; a simplified production process; elimination of the risk of viral contaminations from mammalian components; and intellectual property advantages.
We developed ProCellEx based on our plant cell culture technology for the development, expression and manufacture of recombinant proteins which are the essential foundation of modern biotechnology. We develop new, recombinant therapeutic proteins by using the natural capability of agrobacterium to transfer a DNA fragment into the plant chromosome, allowing the genome of the plant cell to code for specific proteins of interest. The agrobacterium-mediated transformed cells are then able to produce specific proteins, which are extracted and purified and can be used as therapies to treat a variety of diseases.
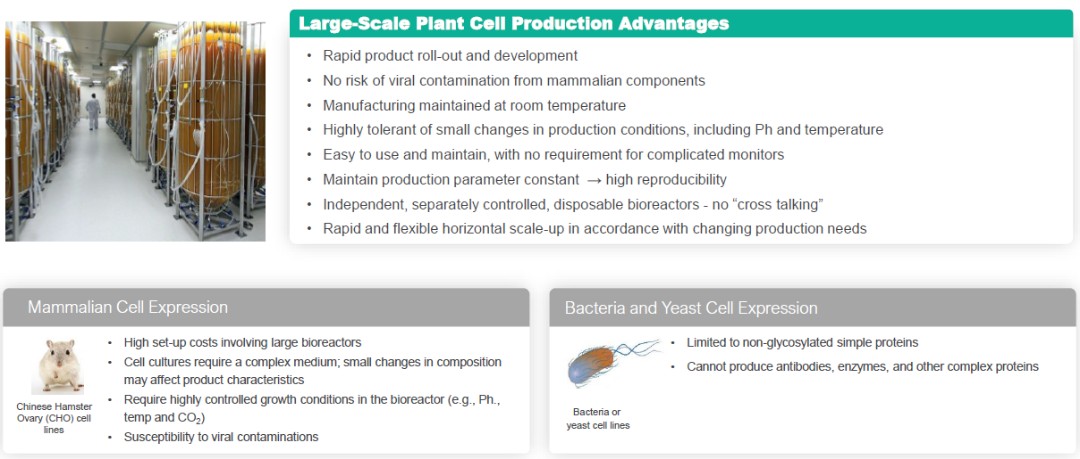
Our ProCellEx technology can be utilized to express complex therapeutic proteins belonging to different drug classes, such as enzymes, monoclonal antibodies, hormones, cytokines and vaccines. The entire protein expression process, from initial nucleotide cloning to large-scale production of the protein product, occurs under Current Good Manufacturing Practice-, or cGMP-, compliant, controlled processes. Our plant cell culture technology uses cells, such as carrot and tobacco (BY-2) cells, which undergo advanced genetic engineering and/or chemical modifications, and are grown on an industrial scale in a disposable, flexible bioreactor system. Our system does not involve mammalian or animal-derived components or transgenic field-grown or whole plants at any point in the production process.
Cell growth, from initiating scale-up steps from a cell-bank through large-scale production takes place in a clean-room environment in flexible, sterile, custom-designed polyethylene bioreactors, and does not require the use of large stainless-steel bioreactors commonly used in mammalian-based systems for recombinant protein production. The ProCellEx reactors are easy to use and maintain, allow for rapid horizontal scale-up and do not involve the risk of mammalian viral contamination. Our bioreactors are well-suited for plant cell growth using a simple, inexpensive, chemically defined growth medium. The reactors, which are custom-designed and optimized for plant cell cultures, require low initial capital investment and are rapidly scalable at a low cost.
Plant Cell Production Advantages
17