in the retina in several cell types including the retinal pigment epithelial cells, or RPE, photoreceptors and retinal ganglion cells. We believe that an S2R antagonist, such as CT1812, may help to regulate the damage-response processes related to these cells that are impaired in dry AMD. After the completion of our ongoing preclinical studies and subject to discussion with the FDA, we intend to advance directly into a Phase 2 clinical trial in the second half of 2022, leveraging our knowledge of CT1812’s preclinical and clinical profile to date. We have initiated discussions with the FDA regarding our plan.
In addition, we are developing other product candidates in the area of synucleinopathies. Synucleinopathies are a group of degenerative diseases characterized by the abnormal accumulation of the alpha-synuclein protein in neural cell bodies, including Parkinson’s disease, or PD, and dementia with Lewy bodies, or DLB.
Our Pipeline
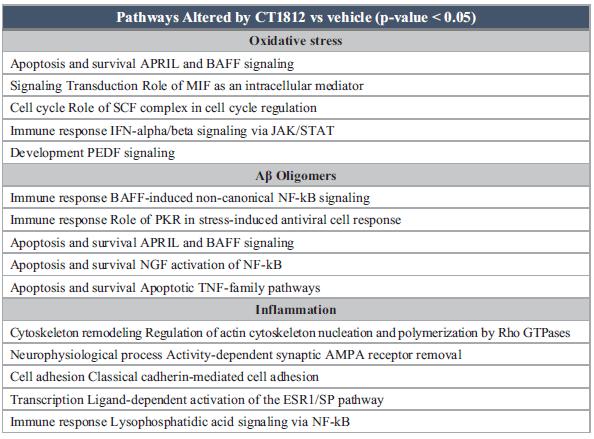
We are developing a pipeline of innovative, small molecule product candidates that are designed to target the S2R complex, a key regulator of the cellular damage response for diseases such as AD, dry AMD, geographic atrophy (an advanced form of dry AMD), or GA, and other conditions for which there is significant unmet medical need. Our current pipeline is summarized below:

Mild to Moderate AD
We are currently engaged in two ongoing Phase 2 clinical trials, designed to evaluate safety, dosing and potential efficacy for CT1812 as a treatment for mild-to-moderate AD. These trials include evaluations of CT1812’s ability to engage with the S2R complex enabling the displacement of Aβ oligomers, its impact in synaptic density and its restoration of synaptic function. In the largest of these trials, our COG0201 SHINE study, we are assessing CT1812’s ability to alter disease progression and cognition, with a target enrollment of 120 participants.
Early-stage AD
We plan to evaluate CT1812 in a 540-patient Phase 2 COG0203 clinical trial to investigate the potential for CT1812’s use at an earlier stage of AD. In addition to cognitive and functional measures, such as CDR-SB, ADAS-Cog and volumetric magnetic resonance imaging, or vMRI, we intend to use a variety of biomarkers to measure target and/or pathway engagement and assess changes in neurodegeneration and disease progression. We will be enrolling patients in the second half of 2022. This trial has been funded by a grant of approximately $81.0 million from the NIA.
7